This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Mutations in the autism-linked gene DNMT3A lead to the behaviors and gene-expression changes seen in different neurodevelopmental conditions.
Mutations in the gene DNMT3A disrupt how other genes are turned on and off, and can lead to a range of neurodevelopmental conditions, a study in mice has found.
The results hint at how mutations in DNMT3A, seen in people with autism and a related condition called Tatton-Brown Rahman syndrome, interfere with brain function, says lead investigator Harrison Gabel, assistant professor of neuroscience at Washington University in St. Louis, Missouri.
DNMT3A codes for a protein that deposits a chemical tag called a methyl group onto the DNA base cytosine. The protein targets only those cytosine molecules that are followed by another base, called adenine, in a DNA sequence. This type of chemical modification, called ‘CA methylation,’ occurs primarily after birth in neurons and accumulates during the first 18 months of life, when neurons are forming connections with other cells, Gabel says.
The protein missing in Rett syndrome — a rare condition characterized by language deficits and repetitive behavior — typically binds to CA methylation sites. So Gabel and his team hypothesized that disrupting the activity of DNMT3A would affect the same cellular mechanisms as those involved in Rett syndrome.
Mice with mutations in DNMT3A show changes in gene expression that mirror those observed in mouse models of Rett syndrome, they found.
“Looking for shared underlying pathology is really important, so that we can start to think about therapeutic approaches that can ameliorate the effects of more than one neurodevelopmental condition,” Gabel says.
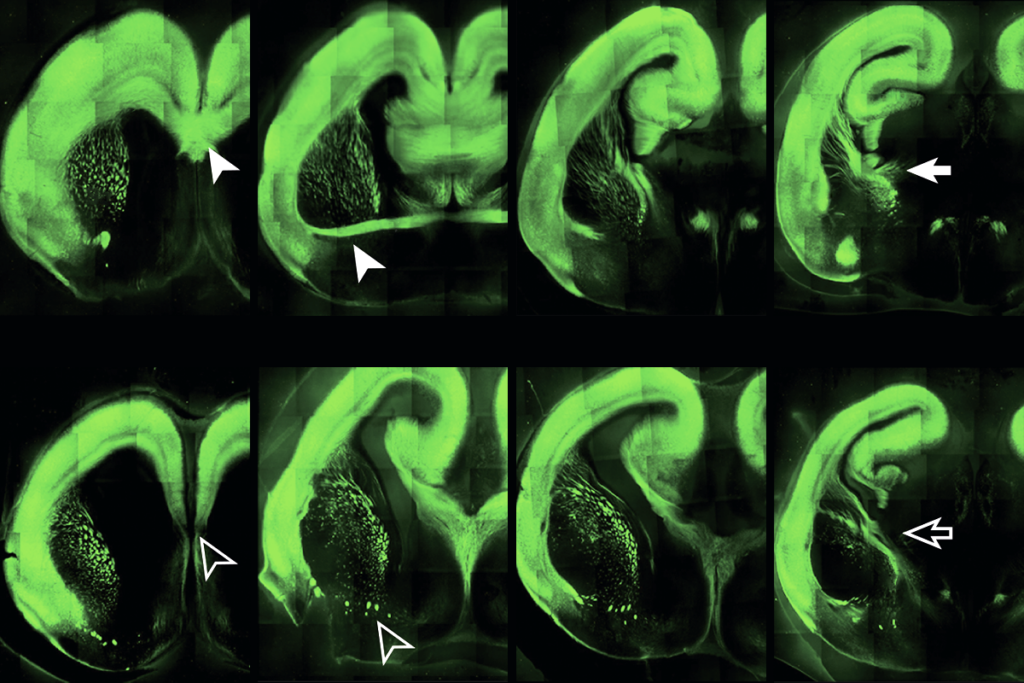
The researchers assessed the effects of nine DNMT3A mutations on DNA methylation in cultured neurons. All nine blocked methylation at CA sites. Mutations associated with intellectual disability had stronger effects than those associated with autism, they found.
The team then deleted one copy of DNMT3A from mice and found that the rodents were heavier and had longer bones, on average, than control mice. These physical features are similar to the overgrowth seen in people with Tatton-Brown Rahman syndrome.
The mice lacking DNMT3A also showed social deficits and other behaviors that resemble core autism traits: The mice spent less time than controls inspecting a new mouse or a new object, and as pups they squeaked less for their mothers than control mice did.
The mice’s neurons have nearly half the usual amount of CA methylation, the researchers found, especially at enhancers, portions of the genome that can regulate gene expression. As a result, several genes showed altered expression.
Many of these genes are also altered in mouse models of Rett syndrome, which suggests that some traits in people with DNMT3A mutations arise from cellular changes like those in people with Rett syndrome, Gabel says. The study was published in November in Cell Reports.
The findings also suggest that CA methylation at enhancers is critical for brain development, and that mutations in DNMT3A contribute to neurodevelopmental conditions, says Magdalena Janecka, assistant professor of psychiatry at the Icahn School of Medicine at Mount Sinai in New York City, who was not involved in the work.
“But this is a study in mice, so it’s a very big leap to say that those mutations are definitely causative of the disease in humans,” Janecka says.
The convergence between mice with one working copy of DNMT3A and mouse models of Rett syndrome is “surprising,” says Annie Vogel Ciernia, assistant professor of biochemistry and molecular biology at the University of British Columbia in Vancouver, Canada, who was not involved in the study. Ciernia notes that mice with one working copy of DNMT3A don’t seem to have the severe motor deficits observed in Rett syndrome mice, perhaps because different types of neurons are affected. But, she adds, “it’s interesting how you can have overlap in the molecular mechanism that leads to very different phenotypic outputs.”
The challenge for researchers is to understand why different mutations can lead to similar features, says Zhaolan Zhou, professor of genetics at the University of Pennsylvania in Philadelphia, who was not involved with the study. Therapeutic applications are years away, Zhou adds, but finding shared features across different conditions could help scientists identify core mechanisms to target.
Next, Gabel and his team plan to assess whether other genes associated with autism affect the methylation at CA sites. They also intend to investigate how CA methylation regulates gene expression in the brain.