This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
A new stem cell model of Angelman syndrome delivers evidence for three molecular mechanisms underlying the condition — and one potential treatment.
A new stem cell model of Angelman syndrome delivers evidence for three molecular mechanisms underlying the condition — and one potential treatment. Researchers presented the unpublished results today at the 2016 Society for Neuroscience annual meeting in San Diego.
The study found that the primary genetic cause of the syndrome, deletion of the UBE3A gene, short-circuits electrical activity in developing neurons. And faulty electrical activity may prevent the cells from forging and maintaining connections, or synapses, with neighboring neurons.
UBE3A is located on a stretch of chromosome 15 called 15q11-13 and is also implicated in autism. Typically, people inherit two copies of the gene and express the maternal copy, while the paternal copy is silenced. But people with Angelman syndrome lack the maternal copy and do not express the gene at all. This produces a cheerful disposition, seizures, intellectual disability and little or no speech.
“To develop therapeutics to target [Angelman syndrome], we need to understand the targets of UBE3A,” says lead investigator Eric Levine, professor of neuroscience at the University of Connecticut in Farmington, Connecticut.
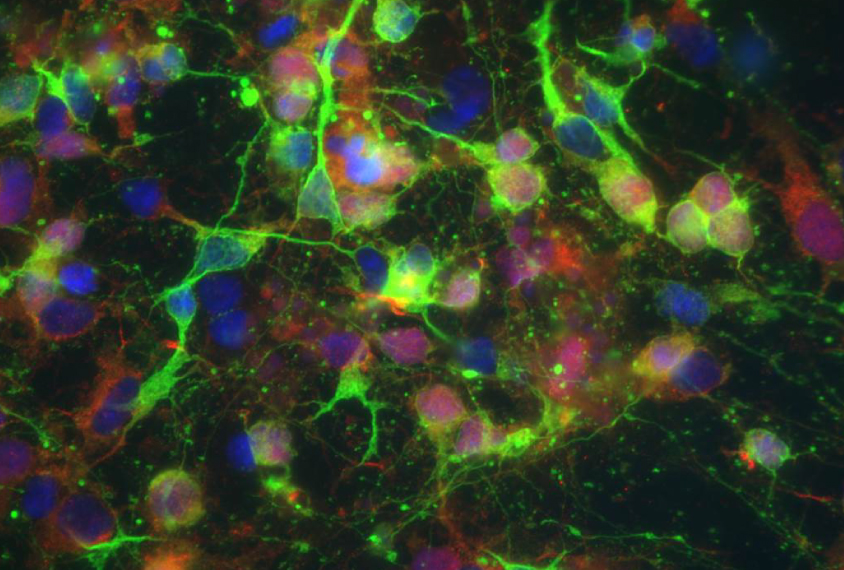
Levine’s team took skin cells from three people with Angelman syndrome and four controls, and derived stem cells from the samples. They induced the stem cells to form a diverse population of neural stem cells, mimicking the mix found in the human brain.
Beginning with the stage that would be present at the onset of embryonic development, the researchers monitored the stem cells’ development, electrical activity and ability to forge synapses with other neurons. They continued to monitor these features for more than 20 weeks, capturing the period of development when synapses begin to function.
The cells that are derived from people with Angelman syndrome and the typical cells appear identical during the first five weeks of embryonic development, the study found. But by week six, the typical stem cells show mature electrical firing patterns and synapse formation, whereas the Angelman cells remain in an immature state.
By 20 weeks, the control cells can strengthen or weaken their synapses, a process known as synaptic plasticity. The Angelman cells do not show this ability.
“At the very earliest stages of development, neurons from patients actually look very similar to neurons from controls,” Levine says. But later in development, he says, “cells from Angelman patients show profound impairment.”
The researchers then explored exactly at what point and how the Angelman cells begin to diverge from controls.
They used the DNA editing system CRISPR to silence UBE3A in control cells at the earliest point of embryonic development. This disrupts all three features they had observed: electrical activity, synapse formation and synaptic plasticity.
By silencing UBE3A at other points in development, the researchers found that electrical activity goes first, and that loss in turn undermines synapse form and function.
To see whether they could reverse the deficits, the researchers treated the Angelman stem cells with a cancer drug called topotecan, which activates the silenced copy of UBE3A. The treatment corrects all three problems.
The results suggest that un-silencing UBE3A early in development helps prevent the chain of events that UBE3A’s loss sets in motion. The findings jibe with other studies showing that Angelman symptoms can be averted if the father’s copy of UBE3A is activated during critical periods of development.
For more reports from the 2016 Society for Neuroscience annual meeting, please click here.