Listen to this story:
0:00
/
The mutations disrupt protein translation as well as the cell’s skeleton, according to a new study.
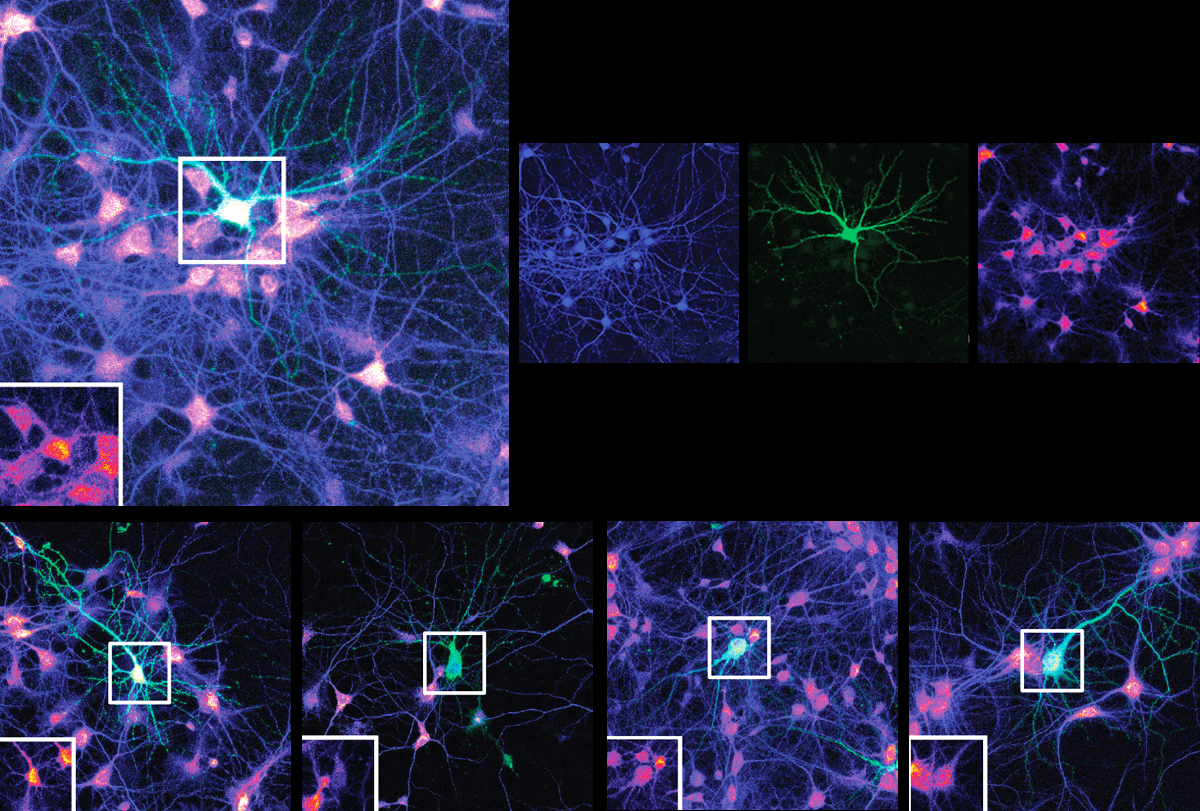
Mutations in an autism-linked gene impede the extension of amino acid chains, the building blocks of proteins, a new study suggests. The findings could help explain why neurons from people with the mutations grow fewer, shorter dendrites, the branch-like structures that receive signals from other cells.
Protein production involves three stages: initiation, elongation and termination. Several forms of autism involve issues with the first stage, a failure of protein synthesis to get going. Problems with protein elongation may also contribute, emerging evidence suggests.
“There’s probably more regulation at the elongation level in neurons than I gave credit to before,” says lead investigator Eric Klann, director of the Center for Neural Science at New York University in New York City.
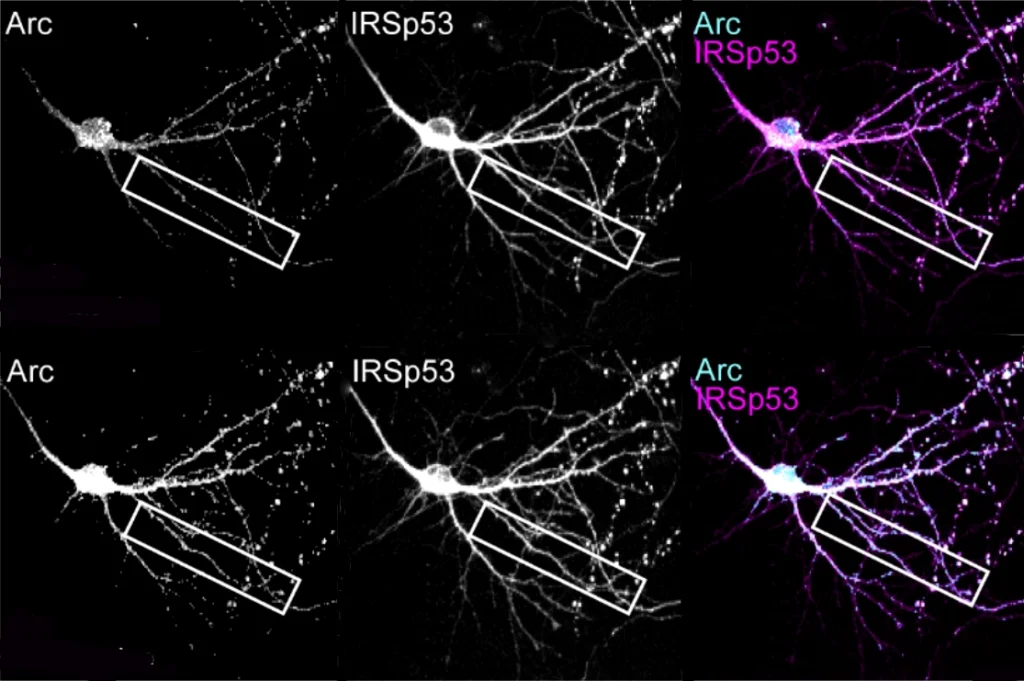
Multiple mutations in EEF1A2, a neuron-specific elongation factor, are associated with autism, epilepsy and neurodevelopmental delay. The protein typically has two functions: transporting molecules called transfer RNAs (tRNA) to the sites of protein synthesis and assembling filaments of actin, a key cytoskeletal protein, into larger bundles.
Unlike the typical protein, mutated EEF1A2 welds itself to tRNAs, the study found. The result is a dearth of tRNA, which in turn hinders the growth of amino acid chains. Like a building abandoned mid-construction, protein creation comes to a halt without these molecules. The mutations also reduce EEF1A2’s ability to bundle actin.
“It’s really good work shedding light on the mechanisms of EEF1A2 mutations,” says Daniel Romaus-Sanjurjo, a postdoctoral fellow at the Health Research Institute of Santiago de Compostela in Spain, who was not involved in the research. The gene, he adds, is becoming a “research hotspot,” because protein synthesis and actin remodeling play a key role in neurodevelopment and cancer.
K
Neurons harboring mutated EEF1A2 grew fewer, shorter dendrites with fewer branches than control cells. In all lines — including cells with two working copies of the gene — overexpression of the mutated proteins dampened protein synthesis. This finding suggests that neural dysfunction is caused by the mutated protein’s toxic effects, rather than a lack of EEF1A2. The findings were published last month in the Proceedings of the National Academy of Sciences.
Protein elongation and actin bundling — another key process in dendrite formation — were disrupted in kidney cells that harbored a mutated protein, the researchers found. “It’s kind of a two-pronged action,” says study investigator Muhaned Mohamed, a graduate student in Klann’s lab.
Either one of these disruptions could lead to neuronal dysfunction, adds Romaus-Sanjurjo. But it’s unclear whether both are at play in explaining the incidence of shorter, sparser dendrites.
“There’s still a lot we need to untangle,” says Froylan Calderón de Anda, head of the Neuronal Development Research Group at the University Medical Center Hamburg-Eppendorf in Germany, who was not involved in the study. “But they are adding pieces to the puzzle. It’s a nice manuscript.”
I
And fewer proteins — combined with alterations to the cell’s cytoskeleton — could impair the ability of synapses to change over time, the researchers say. By coordinating protein synthesis and actin remodeling at dendrites, EEF1A2 promotes synaptic plasticity, as previous research has found. Variants may interfere with that process, which is often diminished in autism.
The new findings point to disruptions in protein synthesis as a “major player in neurodevelopmental disorders,” Calderón de Anda says. More research is needed to “nail down these hypotheses, but it’s a nice starting point, paving the way for more experiments,” he adds.
The researchers are currently using CRISPR to engineer the mutations in stem-cell-derived neurons. By analyzing active ribosomes, the structures in which protein synthesis occurs, they hope to identify specific proteins affected by the mutations. “In our next paper, we’ll have a lot more answers,” Klann says.