The majority of people lacking a functional copy of the SHANK3 gene have both autism and severe intellectual disability, according to a study published 11 June in Molecular Autism1.
The loss of a single copy of SHANK3 is enough to cause Phelan-McDermid syndrome, a disorder known for severe symptoms that can include autism, developmental delay and unusual physical features.
Estimates of the prevalence of autism in the syndrome have varied widely, from 44 percent to 94 percent. Most estimates arise from small numbers of case studies, each using a different measure to evaluate the disorder.
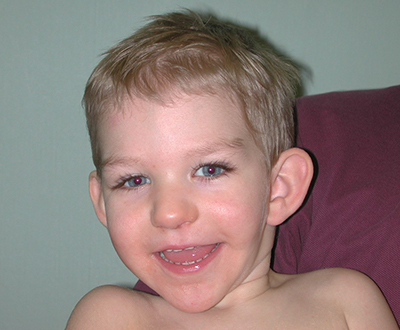
Physical features: A toddler with Phelan-McDermid syndrome shows several distinctive characteristics, including a puffy face, long eyelashes, a broad nose and a pointed chin.
Now, a multidisciplinary team of psychiatrists, psychologists, neurologists, and clinical and molecular geneticists has created a comprehensive picture of the behavioral and cognitive abilities of people who lack a copy of SHANK3.
The researchers found that 84 percent of these people are on the autism spectrum and 76 percent are severely intellectually disabled, with an intelligence quotient below 40. All of them have language problems and 94 percent have difficulty walking.
“Most of the studies to date have relied on parent reports, medical record reviews or on individual case studies,” says lead investigator Alexander Kolevzon, clinical director of the Seaver Autism Center at the Icahn School of Medicine at Mount Sinai in New York.
Deletions and mutations can damage SHANK3. The protein plays a key role in communication across neuronal connections, or synapses. It secures a receptor for a chemical messenger called glutamate to the receiving end of the synapse.
In people with SHANK3 deficiency, “it seems as if, among other things, glutamate signaling across the synapse is compromised,” Kolevzon says. Glutamate signaling is needed to establish learning and memory. Imbalances in glutamate signaling have been implicated in autism.
Precise evaluation:
The researchers evaluated 32 people with SHANK3 mutations referred to them by the nonprofit Phelan-McDermid Syndrome Foundation, other research teams and by word of mouth. The participants were between the ages of 1 and 45, with an average age of 8 years.
Using three assessments — the Diagnostic and Statistical Manual of Mental Disorders IV, the Autism Diagnostic Observation Schedule and the Autism Diagnostic Interview-Revised, the researchers diagnosed 27 of the 32 with either autism or pervasive developmental disorder-not otherwise specified, a condition that includes features of autism.
SHANK3 is located in the 22q13 chromosomal region. The researchers focused on the 30 of 32 people who have deletions in this region and found that the larger the deletion, the more severe the symptoms, including large fleshy hands, seizures and impaired social skills.
This suggests that SHANK3 isn’t the only gene that contributes to the symptoms, says Katy Phelan, director of cytogenetics at Tulane University School of Medicine in New Orleans. Phelan was one of the first researchers to study the syndrome, but was not involved in the new study.
“Most people who have a large deletion in that area are missing 25 to 30 genes near the end of the chromosome,” Phelan says. “We’ve only looked at SHANK3.”
Still, because SHANK3 loss is enough to cause autism, Phelan-McDermid syndrome is a good model for studying autism, notes Guoping Feng, professor of neuroscience at the Massachusetts Institute of Technology, who was not involved in the study. “This is a really good gene to study for the neurological causes of autism.”
Other disorders, such as fragile X syndrome and Rett syndrome, explain 2 percent and 0.5 percent of autism cases, respectively. Phelan-McDermid syndrome causes at least 0.5 percent of autism cases, but the new study suggests the numbers may be higher.
One reason the estimates have been low is that mutations in SHANK3 are harder to find than deletions, because it requires sequencing the entire gene, as opposed to microarray analyses that detect deletions.
“When mutation sequencing is more readily available, it may also be that we find more cases of SHANK3 mutations,” Kolevzon says.
Few studies have assessed people with Phelan-McDermid syndrome at different ages and over time, Phelan adds.
“It’s important to see what happens as these individuals age,” so that families and doctors know what to expect and how to help, she says.
For example, some people with the syndrome lose certain skills as they age, an occurrence known as regression. During the study, five participants lost their ability to speak after they experienced seizures.
In the meantime, the details the researchers gleaned from the study are already proving their worth.
After successful drug tests in mice, the team is conducting the first clinical trial for children with Phelan-McDermid syndrome, looking for improvements in social skills, language and gait. They are testing insulin-like growth factor, a hormone approved for treating short stature that is also in testing as a treatment for Rett syndrome.
Correction: This article has been modified from the original. The previous version incorrectly stated that some of the children were diagnosed with Asperger syndrome.
References:
1: Soorya L. et al. Mol. Autism 4,18 (2013) PubMed