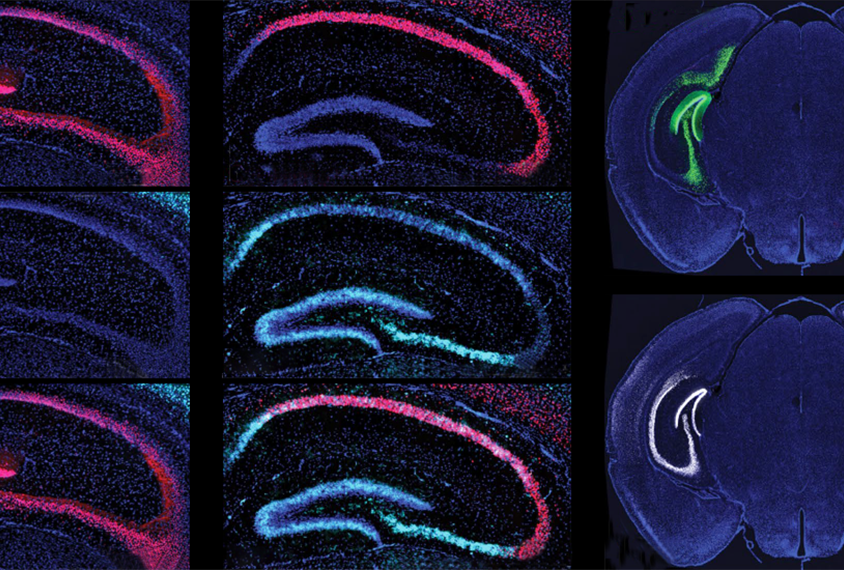
Most cells repair damaged DNA throughout the lifespan while they replicate and divide. But neurons — which stop dividing once they reach maturity and can collect damage every time they send or receive a signal — don’t have that ability.
To help combat the destruction, neurons use a specialized version of a DNA-repair complex called NuA4, according to a study published in Nature on 15 February. Several autism-linked genes encode some parts of that complex, suggesting that disruption to the DNA-repair mechanism may contribute to autism.
The findings help explain how neurons manage what seems to be inevitable damage to their DNA, says Andre Nussenzweig, chief of the Laboratory of Genome Integrity at the National Cancer Institute in Bethesda, Maryland, who was not involved in the work. Neuronal signaling causes double-stranded DNA breaks specifically in promoter regions, which turn genes on in response to brain activity, an independent team showed in 2015.
The new series of mouse experiments shows that neurons elicit NuA4 using NPAS4, a protein that regulates gene expression in neurons in response to brain activity. Some genes that respond to brain activity are strongly linked to autism, according to a 2021 study by the same team.
The NuA4 complex modifies chromatin, the complex of DNA and proteins inside cells. Disruptions to chromatin regulation may sometimes contribute to autism. But the mechanisms by which those regulators work has been unclear, says lead investigator Michael Greenberg, professor of neurobiology at Harvard Medical School. “This work anchors the chromatin regulators that have been implicated in autism to particular pathways and then says, ‘Well, there’s gotta be more of this.’”
G
reenberg and his team had long wanted to understand NPAS4’s role in the brain. They suspected it interacts with many proteins, based on their discovery of a similar complex that centers around FOS, a family of transcription factors that, like NPAS4, are active in the brain immediately after birth. They began by extracting protein complexes containing NPAS4 from the brains of adult mice. The purified molecules weighed about six times what NPAS4 and its known partners should weigh, hinting at more interactions to be discovered.“It was really a pretty key experiment that made the whole project kind of take off,” says study investigator Elizabeth Pollina, assistant professor of developmental biology at Washington University School of Medicine in St. Louis, Missouri.
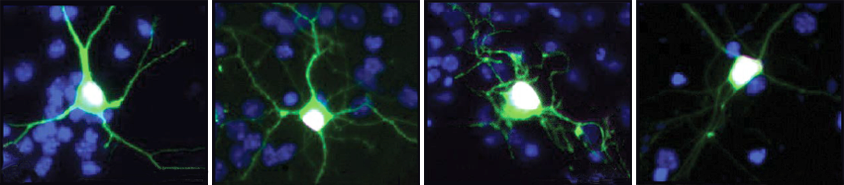
Among those interactions are proteins involved in the NuA4 complex, the researchers found after stimulating neuronal activity in the hippocampus of mice. Some of those genes, including EP400, TRRAP and ACTL6B, have been strongly linked to autism.
That’s not a guarantee that disruptions to DNA repair directly contribute to autism, though, Nussenzweig says. “This complex has other roles.”
Stimulated neurons show an increase in levels of gamma-H2AX — a marker of DNA breakage — in parts of the genome that bind to NPAS4. The damage is centered on more than 1,500 gene regulatory elements, such as promoters and enhancers, according to a method that maps DNA breaks. Ten hours after stimulation, the number of such breaks drops in wildtype neurons, but not in neurons lacking NPAS4 — suggesting that NPAS4 directs the NuA4 complex to regions in need of repair.
A
s mice age, mutations tend to accumulate in parts of the genome not bound by NPAS4, the researchers found. But sites that do bind to the protein seem to be protected from such damage. Mice lacking NPAS4 die at about 1 year old — a year or two earlier than wildtype mice.Neurons accumulate at least three mutations per year, largely at promoters and enhancers, according to a 2022 study by an independent team. The source of the damage seems to be the neurons’ need to rapidly unwind DNA to access genes that help the cells communicate, says Christopher Walsh, professor of pediatrics and neurology at Harvard Medical School and lead investigator on that paper. Walsh collaborates with Greenberg but is not involved in the NPAS4 work.
“These various studies seem to be converging on the fact that yes indeed, when neurons get depolarized, they break open their DNA to turn on their genes very quickly,” Walsh says. “And there has to be a very, very efficient repair process to put it back together again.”
Studying how the NPAS4-NuA4 complex responds to atypical brain activity could clarify how the repair mechanism is linked to neurological and neurodevelopmental conditions, says Daniel Vogt, assistant professor of pediatrics and human development at Michigan State University in Grand Rapids, who was not involved in the work. It will also be important to probe the complex’s role in different subtypes of neurons, Vogt says.
Greenberg says he plans to repeat the work in neurons derived from human stem cells, and to examine the role of the component’s subunits. The work could also help researchers understand the effects of a failure to repair DNA damage over time, Pollina says.
“Now that we have a really nice map of these sites of consistent damage, we’re in a position to ask bigger-picture questions about how that relates to mutational accumulation, particularly in the human brain,” she says.