Listen to this story:
0:00
/
New research is resurfacing old ideas about where the protein forms the disease’s hallmark plaques.
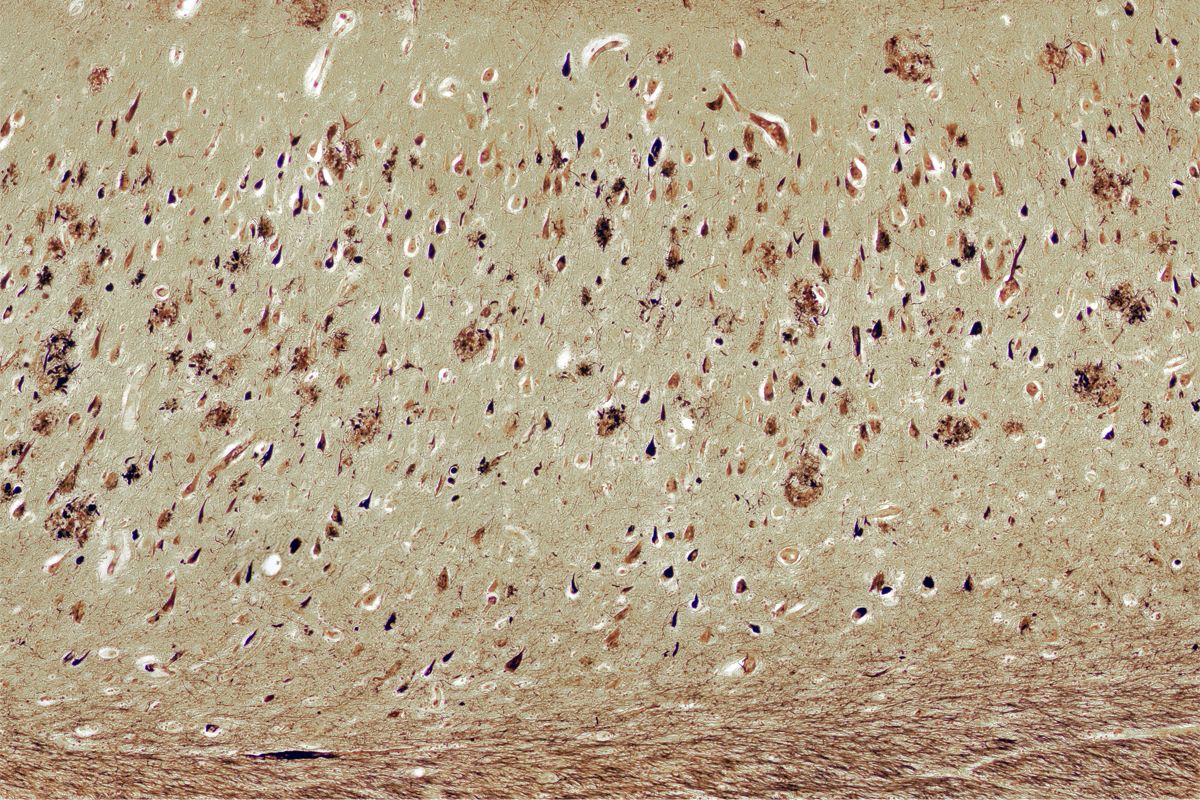
The traditional story of Alzheimer’s disease casts two key proteins in starring roles—each with clear stage directions: Plaques of sticky amyloid beta protein accumulate outside neurons as the condition unfolds, and tangles of tau protein gum up the insides of the cells.
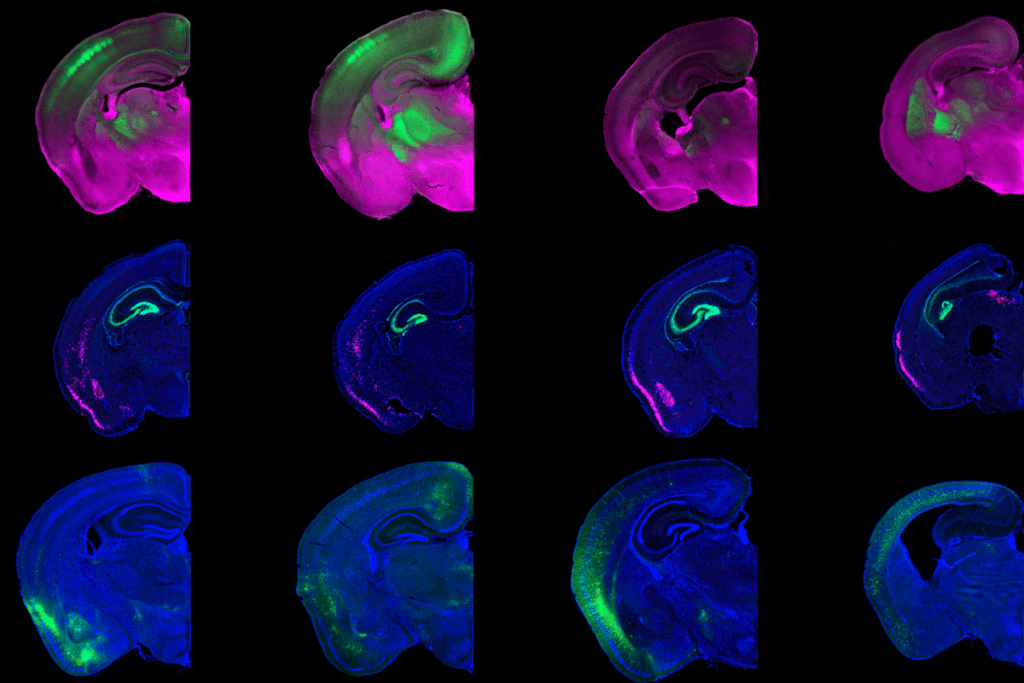
But it may be time for a rewrite. Amyloid beta, too, coalesces inside neurons and seems to mark them for early death, according to research posted on a preprint server last November. In brain slices from people with Alzheimer’s, but not in those from age-matched controls, cells containing intracellular amyloid beta decreased in number as the disease progressed.
At first, the result appeared to be a mistake, says study investigator Alessia Caramello, a postdoctoral researcher in the UK Dementia Research Institute. Intracellular amyloid beta is “nowhere to be found” in most discussions of Alzheimer’s disease, she says. “It’s never mentioned. Never ever.” Instead, the field has long focused on the buildup of amyloid beta outside the cell.
But even before those plaques form, there seems to be another pathological event, she says—namely intracellular amyloid—“Why not look at it?”
The work from Caramello and her colleagues is not the first to suggest that amyloid beta, or Abeta for short, wreaks havoc inside neurons, not just in the extracellular space between them. This “inside-out” hypothesis, as it has been called, has implications for how scientists understand Alzheimer’s disease. In particular, it could help to account for some big mysteries around the condition—such as why the extent of amyloid beta plaques in the brain doesn’t always correlate with symptoms, why neurons die and why treatments to lessen plaques marginally slow down, but do not halt, the disease.
“It just puts a totally different spin on how you need to address this,” says Gunnar Gouras, professor of experimental neurology at Lund University and a proponent of the inside-out hypothesis. “It’s really a cell biological, neurobiological issue that is a bit more complex. And we need to also study this instead of just saying, ‘Abeta is bad; we’ve got to get rid of it.’”
S
everal different research teams say they began studying the accumulation of Abeta within neurons in the decade following 1984, when a team in San Diego first purified the protein. But studies of how it clumps together into the oligomers or fibrils that form intracellular plaques began only in the past decade, says Ralph Nixon, professor of psychiatry and cell biology at New York University.These plaques begin with faulty autolysosomes, the trash collectors of the cell, according to a 2022 study Nixon led. When autolysosomes lose their acidity, they fail to break down proteins such as amyloid beta, and the endoplasmic reticulum surrounding a cell’s nucleus fills with these proteins. Amyloid beta clumps onto itself and forms fibrils within the clogged endoplasmic reticulum and autophagy vesicles. After the neuron dies, it leaves a plaque in its place, Nixon says.
Nixon’s inside-out idea has been slowly gaining traction, and he presented it at a National Academies workshop on Alzheimer’s disease in January. “I think Randy Nixon has really been plowing a lonely furrow and has actually got a lot of it right, for sure,” says John Hardy, professor and chair of molecular biology of neurological disease at University of College London and one of Caramello’s postdoctoral advisers. Hardy introduced the amyloid cascade hypothesis that Abeta plaques cause Alzheimer’s disease in 1992.
As a new proponent of the inside-out hypothesis, Hardy says the classic textbook diagram of how amyloid beta forms—drifting away from a typically unlabeled membrane once cleaved from its parent amyloid precursor protein—is misleading. “Why do we draw Abeta magically leaving the membrane?” he says.
Instead, Abeta is more likely to remain stuck within a membrane—either that of the endosomal lysosome, as Nixon’s work indicates, or the neuronal membrane, Hardy says. Not only is amyloid beta in the part of the amyloid precursor protein that embeds within the membrane, but because it is hydrophobic, he adds, it’s also unlikely to float away into the watery cytoplasm or outside the cell.
The “broad strokes” of Hardy’s amyloid cascade hypothesis hold under the inside-out hypothesis, he says: Regardless of its location, amyloid buildup drives tangle formation; people with early-onset Alzheimer’s disease produce too much amyloid, and people with late-onset Alzheimer’s disease cannot clear amyloid away. “In other words, those of us who are not as good at responding to amyloid deposition are more likely to get disease—basically, that’s the outcome,” he says.
D
espite prominent converts to the inside-out hypothesis, such as Hardy, the idea that that amyloid beta plaques begin outside neurons maintains a loyal following.Amyloid beta-42—the primary form of the protein found in amyloid beta plaques and so named for its 42 amino acids—has too few amino acids to hold it in place in a membrane, says Dennis Selkoe, professor of neurologic diseases at Harvard Medical School. Instead, he argues, Abeta-42 is likely released into the lumen of a vesicle or extracellular space when the vesicle fuses with the cell membrane.
That extracellular space is cramped with cells and vesicles, Selkoe adds. “It’s not like it’s out there in a bathtub of water that’s far away from the wall.” When plaques form outside the cell, clumps of extracellular Abeta-42 might bind back to the outside of membranes and cause local injury to membranes, he says. “That’s really the fundamental mechanism by which many of us think Abeta causes trouble.”
Whether plaques form inside or outside cells—or perhaps involve mechanisms in both locations—the therapeutic approach should remain the same, says Selkoe, who adds that he has advised several companies on Alzheimer’s treatments. “You’d still want to lower the amyloid, regardless of whether you sort of win the argument that it’s intracellular or you win the argument that it’s extracellular.”
But if, instead, plaques are “tombstone markers” of dead neurons, as Nixon’s 2022 paper indicates, the strategy changes, says Charles Glabe, professor of molecular biology and biochemistry at University of California, Irvine. Glabe published a study in 2014 that showed that intracellular Abeta fibrils accumulate within the nucleus of neurons in mice, initiating amyloid plaques.
“Is removing plaques going to do anything?” Glabe asks. “I don’t think so. It’s like taking out the trash after your house burns down—not going to help your living situation.”
A
nd there is another possibility: Plaques could originate in synaptic terminals, says Gouras, who first presented his theory of intracellular Abeta at the Alzheimer’s Association International Conference in 1998 and again 26 years later at the Alzheimer’s & Parkinson’s Diseases Conference in March.If so, that could make some plaques the tombstones of dead neurons, if the terminals are near the neuron’s cell body, as in Nixon’s work. Or, plaques could form in terminals located farther from the cell body, Gouras adds, which could explain why plaques first appear in the cortex but tau tangles first appear in the cell bodies located in the entorhinal cortex, according to a 2021 study he led.
“There’s no one set, completely proven hypothesis,” says Cynthia Lemere, professor of neurology at Brigham and Women’s Hospital, who studies amyloid beta plaques in Down syndrome. She says she thinks plaques begin outside the neuron—and that it is the immune response to the plaque, not the plaque itself, that causes neuronal death.
Other lines of inquiry implicate the immune system as well. For example, nearly all of the risk genes to emerge from a genome-wide association study of late-onset Alzheimer’s disease related to microglia (the primary immune cells of the central nervous system), lipid metabolism genes or both. And that result aligns with Hardy’s theory that membrane-bound amyloid beta attracts microglia to clear up the damage. Many of these same genes are also “amyloid response genes,” whose RNA production increases as amyloid levels go up, according to a 2019 study led by Hardy.
The rewrite of the amyloid plaque origin story ultimately might not matter, Hardy says. As the debates continue, the priority should be to unravel the processes that kill neurons: “We really need to understand why neurons die. I think that fundamentally we need to understand that, not just for Alzheimer’s disease, but we need to understand it as a general principle.”