-

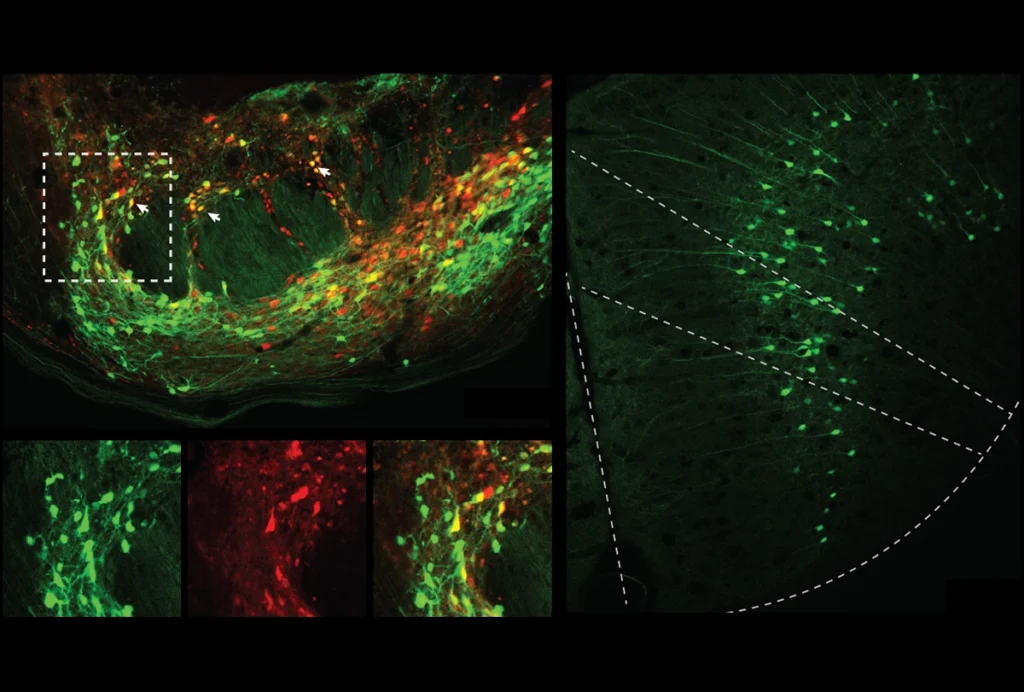
Lost function: Adult mice that lose expression of MeCP2 (green) develop symptoms of Rett syndrome.
-
Lost function: Adult mice that lose expression of MeCP2 (green) develop symptoms of Rett syndrome.
A new study calls into question the assumption that Rett syndrome is exclusively a neurodevelopmental disorder caused by the lack of a critical protein in utero.
Published online 2 June in Science, the study reports that adult mice deprived of MeCP2 show symptoms of Rett syndrome, even though their brains are fully formed and functioning normally before the protein is depleted1.
“I think it’s safe to say that Rett is a postnatal neurological disorder,” says lead investigator Huda Zoghbi, director of the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital in Houston. “It really begs the question whether many other disorders we’ve thought of as neurodevelopmental disorders are really childhood disorders.”
Symptoms of Rett syndrome, which include intellectual disability, seizures and breathing problems, normally appear between 6 to 18 months of age. Children with Rett syndrome often show autism-like behaviors, and the syndrome is considered part of the autism spectrum.
The disorder is usually seen in girls because the MeCP2 gene lies on the X chromosome: Girls have two copies of the gene, whereas boys have just one. Males with the most severe forms of the mutation are either stillborn or do not survive infancy.
In 1999, Zoghbi and her colleagues showed that Rett syndrome is caused by loss of MeCP2, short for methyl CpG binding protein 2, during development2.
In 2007, Adrian Bird and his colleagues reversed many Rett symptoms in mice that carry mutated MeCP2 by restoring the protein in adulthood3. This study gave the first indication that the damage caused by insufficient MeCP2 during development is not irreversible. In retrospect, it also provided the first hint that Rett is not solely a disorder of development.
“The writing was on the wall after our experiments,” says Bird, professor of genetics at the University of Edinburgh in Scotland, who was not involved with the new work.
“There’s been this idea that once you have neurological deficits, it’s too late to do anything about them,” he says. “I think this is an assumption that’s been accepted without experimental evidence. One has to question those assumptions now.”
Other researchers urge caution in interpreting the study’s findings, however.
“I don’t think that these interesting results would argue that MeCP2 is not important in neurodevelopment or that Rett is not a neurodevelopmental disorder,” says Janine LaSalle, professor of medical microbiology and immunology at the University of California, Davis. “They instead show that there is a lifelong requirement for MeCP2.”
Adult onset:
In the new study, Zoghbi and colleagues created a conditional mutant, blocking MeCP2 expression in 60-day old mice after allowing them to develop normally. Within weeks, the adult mice develop gait abnormalities similar to those of young mutant mice that never expressed the protein. The adults also display abnormal nesting behavior and impaired learning and memory, reminiscent of the juvenile mutants.
“We predicted a hybrid that would have some of the symptoms, but in a milder form,” Zoghbi says. Instead, the animals behaved as if they had lacked the protein all of their lives. “That unequivocally told us that neurons constantly need the protein to function properly,” says Zoghbi.
The adult mice die 10 to 13 weeks after the protein is blocked, roughly equivalent to the lifespan of mice that lack MeCP2.
It’s not surprising that MeCP2 is needed for mature neuronal function, LaSalle says. “However, death was a somewhat surprising endpoint.” The mice in the new study should be compared with those from Bird’s study, she says.
MeCP2 is believed to fine-tune the regulation of thousands of other genes. The new study shows that MeCP2 does not affect the hardwiring of brain function because the animals were adults and “hardwired normally,” says Zoghbi. “It’s more the software.”
This is supported by results from another study, published in April, which showed that even in the absence of MeCP2, brain circuits appear to form normally4.
The new study suggests that any therapies for Rett syndrome that rely on replacing the missing MeCP2 protein must be continuously maintained. This may also be true for proteins implicated in other neurodevelopmental disorders. “You can’t administer it just once,” says Bird. “This is a protein you are going to need on a continuous basis.”
Correction: This article has been modified from the original. A quote containing a factual error about the results has been deleted.
References: