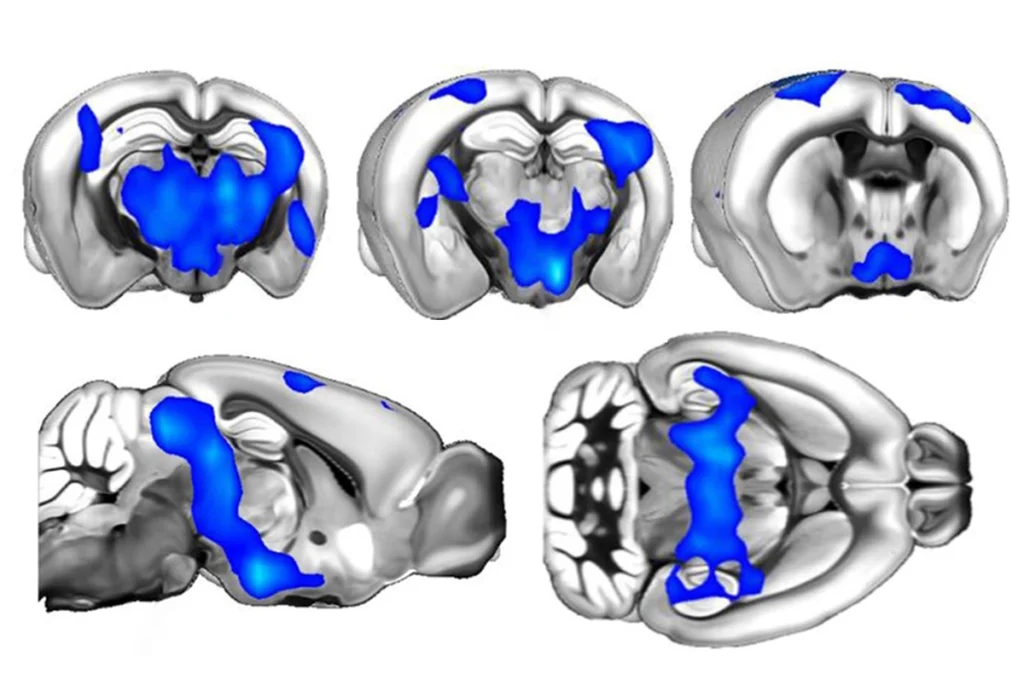
The genetic underpinnings of autism are complex and hard to parse in most cases. Scientists have identified at least 100 genes that contribute to autism. But there are a few rare syndromes in which a single mutation can lead to the social and behavioral characteristics of autism.
For scientists, the utility of these conditions is that they allow us to study how a single malfunctioning gene leads to autism-like social behaviors. My lab focuses on two of these: Smith-Magenis and Potocki-Lupski syndromes. Both stem from a problem in a gene called retinoic acid induced 1 (RAI1). Smith-Magenis syndrome results when one copy of RAI1 is missing; Potocki-Lupski syndrome occurs when a person has three copies of the gene.
Each syndrome comes with distinctive facial features and a constellation of medical issues, but what is of particular import to our research is that they both share traits of autism — notably, trouble reading social cues. People with these syndromes are typically extremely trusting, treating strangers with the same familiarity as family. For example, those with Smith-Magenis are often unusually affectionate, and those with Potocki-Lupski are unusually chatty.
The precise function of RAI1 in the brain remains unclear, but we are chipping away at the mystery. First of all, we know that RAI1 produces a transcription factor — that is, a protein that mediates gene expression. If the human genome is a workplace, transcription factors are the high-level managers. These proteins act on multiple other genes, dictating when and how much they are active, and in which cells of the body.
A mutation in RAI1 causes a ripple effect, therefore, dysregulating hundreds of other genes under its control. This is why Smith-Magenis and Potocki-Lupski syndromes include such disparate physical and cognitive problems, including kidney dysfunction, sleep apnea, scoliosis, and vision and hearing loss, along with behavioral and learning issues.
Our lab focuses specifically on the social challenges people with these syndromes face, which are presumably caused by autism-related genes within the RAI1 network.
Awkward mice:
By unraveling the molecular activities of the RAI1 protein, we hope to learn how to activate or mute it, thereby eliminating unwanted medical and cognitive problems. Mice whose RAI1 genes have been altered or removed show distinct behavioral abnormalities, reminiscent of people with the syndromes.
Typical mice are sociable creatures. They prefer spending time in one another’s company over being alone or with an inanimate object. They also show a preference for ‘social novelty’ — meaning that, given the choice of approaching a mouse they know or one they have never met, they will seek to make the acquaintance of the stranger.
Mice with altered RAI1 genes are as sociable as typical mice, but they are indiscriminately sociable, evincing no preference for social novelty1. When given the option of exploring either a space with an object or one containing another mouse, they approach the mouse, like controls do. But when one of the two spaces contains a familiar animal and the other a newcomer, the RAI1 mice show no preference. This inability to distinguish between friend and stranger (or lack of interest in doing so) is one of the key autism traits seen in people with Smith-Magenis and Potocki-Lupski syndromes.
We also gave the mutant mice a test that is normally used to assess aggression. Control mice retreat in this test as often as they advance. But Potocki-Lupski mice win practically every round, pushing forward insistently as the other mice back their way out2. We don’t think this behavior reflects aggressiveness, but rather a single-minded attachment to their goals, coupled with a lack of awareness of the other mouse’s unease. Smith-Magenis mice, on the other hand, retreat almost every time3.
Why do mice with extra RAI1 reliably push through the tube, whereas those lacking some or all of the gene retreat? We do not know, but this difference suggests that the amount of the gene that is present is significant in determining the form of abnormal behavior.
Big winners:
A few years ago, our lab created mice that express an excess of RAI1 in their forebrain neurons. These Potocki-Lupski mice are less able than controls to learn and remember new things. We gave these mice the antibiotic doxycycline, which restores normal levels of gene expression. Disappointingly, we saw no improvement in their memory or learning4.
However, last year, another team published results suggesting that it is possible to reverse RAI1-related behavioral abnormalities, but only during a specific time in a mouse’s life. The researchers created Smith-Magenis-like mice and normalized RAI1 levels at either 3 weeks of age (the equivalent of a teenager) or 7 weeks (adulthood).
The mice treated as adults continued to be big losers in the tube test. But those treated at 3 weeks won half their face-offs with controls and beat other Smith-Magenis mice a full 86 percent of the time5.
I find these results extremely hopeful. They suggest that eventually we can find strategies to ease or even reverse autism traits in people with these rare genetic conditions. And from that research may come therapies that work for a broader group of people with autism.
Katherina Walz is research associate professor at the Hussman Institute for Human Genomics at the University of Miami in Florida.