This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Autism may be the result of faulty wiring that occurs during early brain development, according to two independent studies that looked at the origins of circuit disruption.
Autism may be the result of faulty wiring that occurs during early brain development, according to two independent studies that looked at the origins of circuit disruption.
Both teams found defects in a particular type of interneuron — intermediary cells that link sensory neurons to motor neurons — in the cortexes of mice.
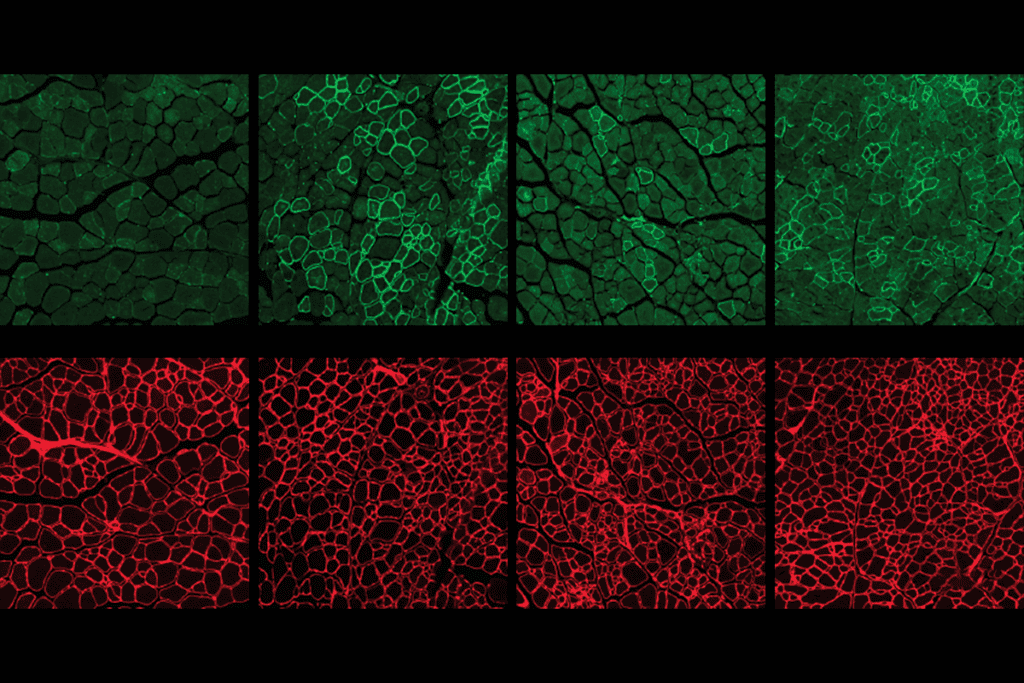
In the first paper, published in June in the Journal of Neurodevelopmental Disorders, Takao Hensch and his colleagues investigated a subset of interneurons — those that contain the calcium-binding protein parvalbumin — and found that there are fewer of those cells in the cortexes of nine different mouse models of autism1.
A second paper in the 22 September Proceedings of the National Academy of Sciences reported that decreased function of a section of genes on chromosome 22 results in the abnormal placement of parvalbumin interneurons and alters the development of the cortex2.
Interneurons trigger the release of neurotransmitters, chemicals that facilitate nerve cell communication. Inhibitory neurotransmitters control the ‘off’ switch in a nervous system circuit, whereas excitatory neurotransmitters turn on the circuit.
Parvalbumin-positive inhibitory neurons make up 40% of the gamma-aminobutyric acid (GABA) cell population, the cells that dictate the timing of brain development by tempering the overwhelming excitatory input to the brain3.
Together, the papers add heft to the hypothesis that an imbalance between excitatory and inhibitory neurons underpins autism. The high prevalence of seizures in individuals with autism is seen an indicator that the balance between excitatory and inhibitory circuits has gone awry in people with the disorder.
“The possibility is emerging that subtle changes in the numbers and proportions of this category of interneurons may result in a range of neurodevelopmental disorders, including schizophrenia, bipolar disorder and autism,” says Anthony-Samuel LaMantia, lead investigator on the PNAS study and a neuroscientist at the University of North Carolina at Chapel Hill.
The studies also emphasize the essential role inhibitory interneurons play in the development of proper circuitry in the cortex. Restoring circuit balance may therefore be a plausible way to reverse autism, suggests Hensch, a neurobiologist at Harvard University.
Hensch and his colleagues discovered 12 years ago that a certain amount of GABA function is needed for the brain to develop correctly. Research from his and other labs showed that the parvalbumin cells dictate the timing of circuit maturation.
Because autism is a neurodevelopmental disorder that appears to result from altered trajectories of brain development, Hensch’s team decided to search the literature for a common circuit defect in mouse models of autism.
Hensch’s previous work had shown that a balance of excitatory to inhibitory activity is needed in order for a brain circuit to develop correctly. “But no one had systematically looked at subtypes of circuits that may be defective across autism models,” he says. “We were struck to find that all these papers report defects in parvalbumin-containing cells.”
Hensch’s team then studied two additional mouse models of autism in detail: One model is the result of an embryonic chemical insult from the antiepileptic drug valproate, and the other the result of a single-gene mutation of neuroligin-3, a protein that mediates neuronal communication.
The researchers found that mice exposed to valproate have very few to no parvalbumin cells in the neocortex compared with controls. The abnormal zones span several hundred micrometers, but are surrounded by areas with normal numbers of parvalbumin cells.
Intriguingly, the abnormal regions appear to affect only one hemisphere of the neocortex, which, the researchers say, is indicative of a strong imbalance in the electrical circuit — one that would favor inhibition over excitation between the two sides of the brain.
The team found similar defects in the neuroligin-3 mouse model.
“I think [parvalbumin] is a sensitive circuit-level marker of neurodevelopmental disorders, including autism and schizophrenia,” Hensch says.
Impairment of these interneurons could have two major developmental effects, both of which come down to timing, Hensch notes.
First, parvalbumin cells can accelerate or delay critical periods of brain development or failure. The cells also generate rhythms known as gamma oscillations that, if dysregulated, could result in the gross mistiming of a circuit’s response to sensory input.
DiGeorge syndrome is a congenital disease that results from the deletion of roughly 30 genes in the middle of chromosome 22, a region dubbed 22q11. Mouse models of this syndrome are unique because they can precisely parallel the same genetic defect that, in humans, heightens the risk of schizophrenia and autism.
LaMantia’s team found that six of the deleted 22q11 genes are normally expressed in the earliest developing cortical cells, and their loss may alter cortical development. Deletion of the whole set of genes disrupts the proliferation of what will become the earliest cortical cells and leads to abnormal placement of parvalbumin interneurons in the upper and lower layers of the developing cortex.
Interestingly, says LaMantia, a number of neurodevelopmental disorders are marked by genetic changes during cortex development, but manifest themselves through behavior only later in life.
“If you put all of the disorders of cortical connectivity from schizophrenia to bipolar disorder to autism in a three-dimensional space, underneath all of them would be changes in two mechanisms: the neurogenesis, migration and placement of cortical neurons, and the genesis of GABA-producing interneurons,” says LaMantia.
Although several research groups are focusing on the same population of parvalbumin cells, they are not seeing changes of the same magnitude or type, LaMantia notes. For example, LaMantia says he did not find a difference in the number of parvalbumin interneurons, or in their presence in the two hemispheres of the brain.
That may be because La Mantia’s study examined large deletions that affect multiple diverse genes, which may cause irreversible defects, counters Hensch. “We focused on the simplest cases, the result of single-gene mutations, to glean the principles of circuit disruption,” Hensch says.
Still, the papers together signal a shift in focus from gene hunting to circuit function, says Hensch.
One specific function of the parvalbumin cells — generating and synchronizing gamma oscillations in the hippocampus and neocortex — may well prove to be a robust biomarker of neurodevelopmental disorders, Hensch says.
Gamma oscillations are a pattern of brain waves detectable by electroencephalograms, and are thought to control the timing of the brain’s ability to process information. Weak or altered gamma oscillations have been found in people with autism and schizophrenia.
The highly distributed processing of sensory information in the cortex requires nerve cells to be synchronized. The GABA network is a type of pacemaker for these cells, without which the brain develops faulty connections, says Wolf Singer, a neurophysiologist at the Max Planck Institute for Brain Research in Frankfurt, Germany.
“A breakdown in the GABA network may explain why the brains of children with autism have difficulty establishing long-range synchrony in high-frequency gamma waves,” Singer says.
Decreased long-range connectivity is central to many current hypotheses of autism’s origins.
Several studies suggest that a lack of connectivity underpins the autism brain’s inability to synchronize across the two hemispheres — and that may be the result of lower inhibitory input4,5. Hensch and his colleagues suggest that this circuit-level imbalance predictably delays the timing of critical periods of brain development and inhibits synchronization across the brain.
In fact, Hensch suggests parvalbumin cell activation underlies the successful genetic manipulation that corrected fragile X syndrome in a mouse model of the disorder6. Restoring the excitatory/inhibitory balance in the circuit may similarly reverse autism, he says.
In the meantime, assessing defects in interneurons is challenging because, at a gross level, the brains of people with these disorders are intact and working — albeit a little differently. “These are going to be very subtle changes in the number and proportion of cell classes,” says LaMantia.
Josh Huang, a neurobiologist at Cold Spring Harbor Laboratory, is developing the tools needed to visualize and manipulate most types of inhibitory neurons.
“We need to be able to disrupt these neurons in ways sufficient to track specific behavior or cognitive deficits,” says Huang. His research focuses on GABA inhibitory cells, including parvalbumin-containing cells, that are essential to the assembly and function of neurocircuits.
“Once we have the tools, we’ll see where and how changes occur in defined functional components of neural circuits,” Huang says, “and what impact that has on neurocircuit development.”