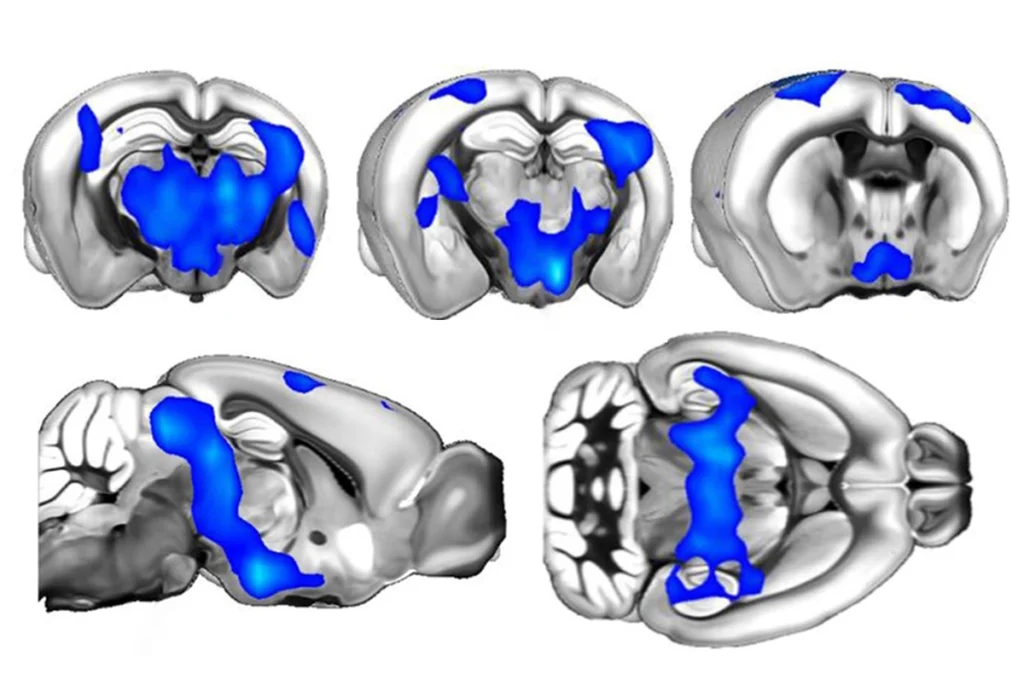
The mutation that causes tuberous sclerosis complex, an autism-related disorder, may disable calming signals in the brain, leading to hyperactive neurons, according to a study published 8 May in Neuron1.
Tuberous sclerosis is caused by mutations in either TSC1 or TSC2 and is characterized by benign tumors, called tubers, throughout the body. One of its predominant symptoms is severe seizures, seen in as much as 90 percent of people with the disorder. The new study relies on mice lacking both copies of TSC1.
Studies have shown that mouse models of the disorder have elevated brain activity, which may be the cause of these seizures. But researchers were not clear on how exactly the TSC mutations trigger the changes.

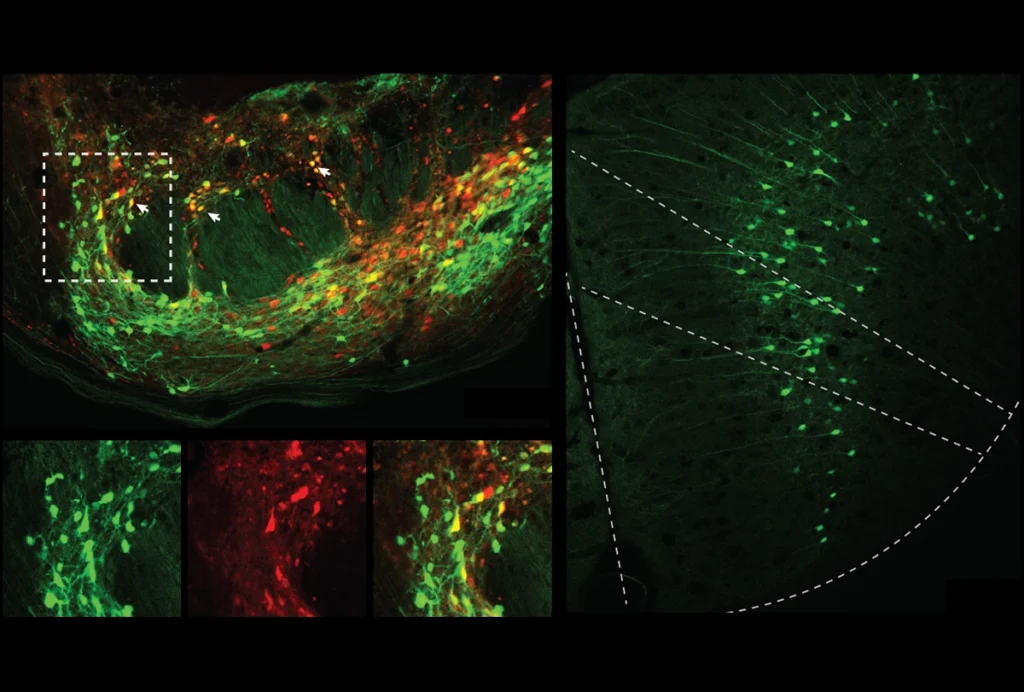
Brain boost: Deleting one of the genes that leads to tuberous sclerosis (left, green) elevates mTOR levels (red) compared with control neurons (right), and may lead to seizures.
“That is the frontier we know jack about,” says Gordon Fishell, professor of neuroscience at New York University, who was not involved in the study. “Despite the overt example of the tubers, I don’t think anyone really had any clear idea of what was happening.”
The popular theory for what causes seizures in tuberous sclerosis is that excitatory neurons, which initiate brain activity, are inherently hyperactive in people with the disorder.
The new study instead found that in the mouse model, the excitatory neurons don’t respond to signals from inhibitory neurons, which dampen their activity. Although the two sets of neurons form connections, or synapses, just as they do in controls, excitatory activity in the mutant mice remains unchecked.
This may be because the mutant excitatory neurons are missing some of the receptors that respond to gamma-aminobutyric acid (GABA), the chemical messenger released by the inhibitory neurons, the researchers say.
“It shows how wrong our intuition was about the system,” says senior investigator Bernardo Sabatini, professor of neurobiology at Harvard Medical School. “We kept going through one thing or the other until we finally had to face the fact that it was inhibition.”
Secondary changes:
The researchers first looked for signs of hyperactivity in cultured neurons lacking both copies of TSC1. They found that the excitatory neurons have altered patterns of signaling and gene expression.
But many of these changes would be expected to dampen, not boost, excitatory activity — for example, the mutant neurons have lower-than-normal levels of GLUA1 and GLUA2, components of a receptor that responds to excitatory signals. Levels of ARC, a protein that helps keep excitatory activity in check, are elevated in these neurons.
These alterations may be an attempt to compensate for the lack of inhibition, the researchers say. “[The excitatory neurons] are actively trying to restore balance to the system, but for some reason it isn’t working,” says Helen Bateup, assistant professor of neurobiology at the University of California, Berkeley. Bateup did the research as a postdoctoral fellow in Sabatini’s lab.
To separate the primary effect of lack of TSC1 from its ultimate influence on brain circuits, the researchers then deleted the gene in a small number of cells in the hippocampus. They activated this mutation in 14- to 17-day-old mature mice after brain circuits had already formed.
In these mice, the mutant excitatory neurons show a weaker response to inhibition than control neurons do, the researchers found.
“This is a novel finding that will open up new avenues for research,” says Mustafa Sahin, assistant professor of neurology at Boston Children’s Hospital, who is not involved in the study. “Why do inhibitory synapses change in those neurons?”
The results also help elucidate how rapamycin, a drug that is in clinical trials for tuberous sclerosis, works to quell seizures. Rapamycin corrects both the deficit in inhibition as well as the gene expression and other changes in the excitatory neurons.
Rapamycin inhibits the activity of mTOR, a protein involved in cell growth that is kept in check by TSC1 and TSC2. The study suggests that mTOR, which is elevated in the mouse model, leads to deficits in inhibition, although how it does this is still unknown.
Rapamycin may also be useful for seizure disorders that are unrelated to mTOR regulation but which show a similar block in inhibition, says Sabatini.
If the hyperactive signaling is the result of missing GABA receptors, as the study suggests, a commonly prescribed group of anti-seizure medications that activate GABA signals would be ineffective, notes Sabatini. Researchers should look for new compounds that may restore these receptors, he says.
About half of individuals with tuberous sclerosis have autism, and researchers have posited that a similar imbalance between excitatory and inhibitory signaling may underlie autism symptoms.
“We are getting to the point where we need to focus on cellular mechanisms,” says Fishell. “That makes this study an important step forward for autism, even if the genetics [of autism and tuberous sclerosis] aren’t tightly associated.”
References:
1: Bateup H.S. et al. Neuron 78, 510-522 (2013) PubMed