Listen to this story:
0:00
/
A protective pathway that pauses protein synthesis is muted in a mouse model of fragile X syndrome, according to a new study.
Fragile X syndrome mutes a key stress response pathway in neurons, according to a new study in mice. And activating the pathway reverses behaviors reminiscent of the condition — pointing to new potential treatment avenues.
Fragile X syndrome, the most common inherited cause of autism, arises from mutations that prevent the synthesis of a protein called FMRP. Mice that lack FMRP overproduce proteins at synapses, disrupting neuronal function. Part of that overproduction in the animals likely reflects overactivity in the mTOR pathway — a signaling cascade that regulates cell division and protein synthesis.
But another pathway controlled by mTOR could also contribute to the excessive protein production, says lead investigator Arkady Khoutorsky, assistant professor of anesthesia at McGill University in Montreal, Canada.
That pathway involves a protein called eIF2-alpha, which governs a cell’s response to stress — a viral infection or nutrient depletion, for example. Such stressors add a phosphate group to eIF2-alpha, inactivating it and stalling protein synthesis so that cells can reallocate resources.
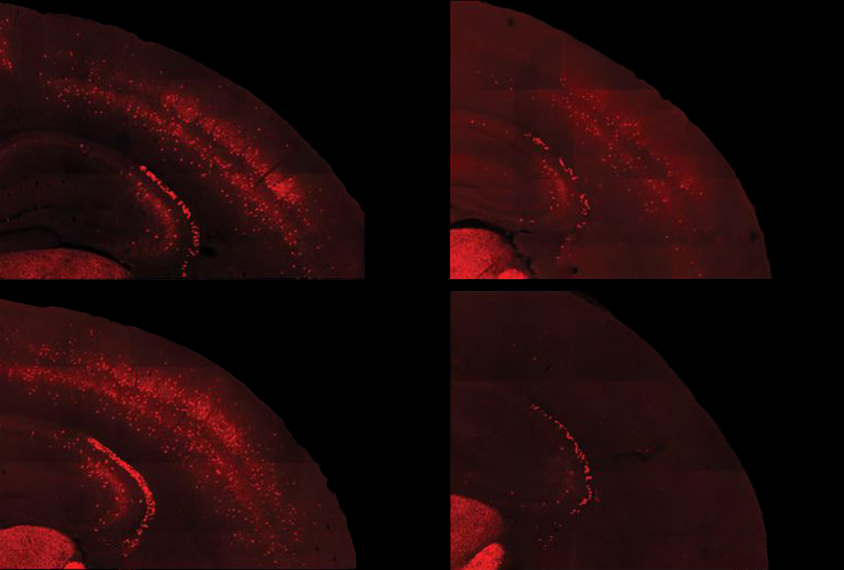
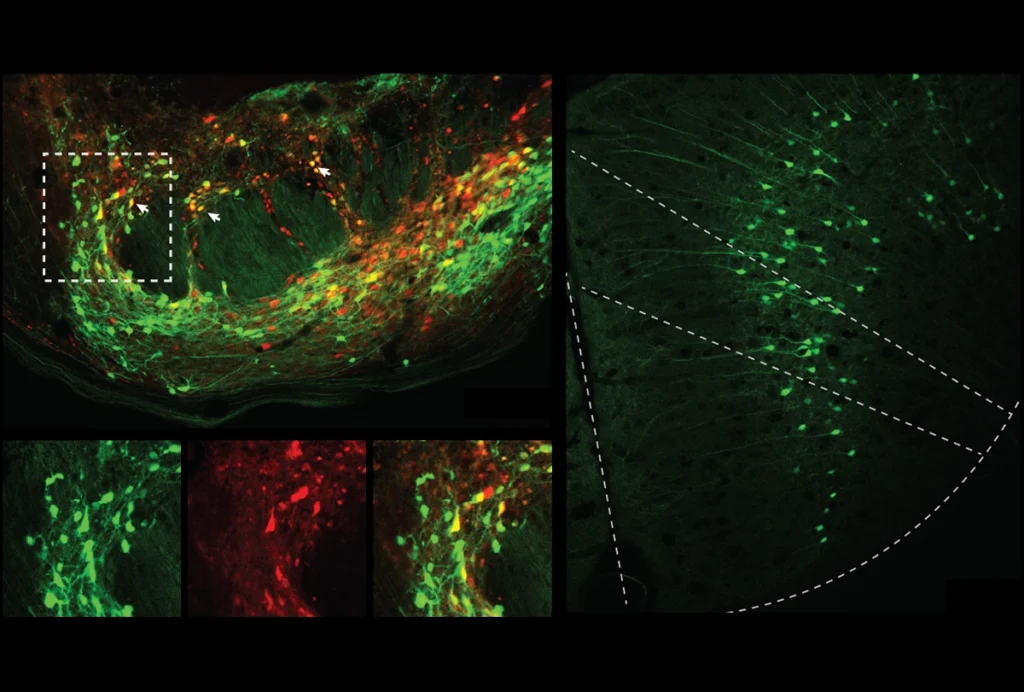
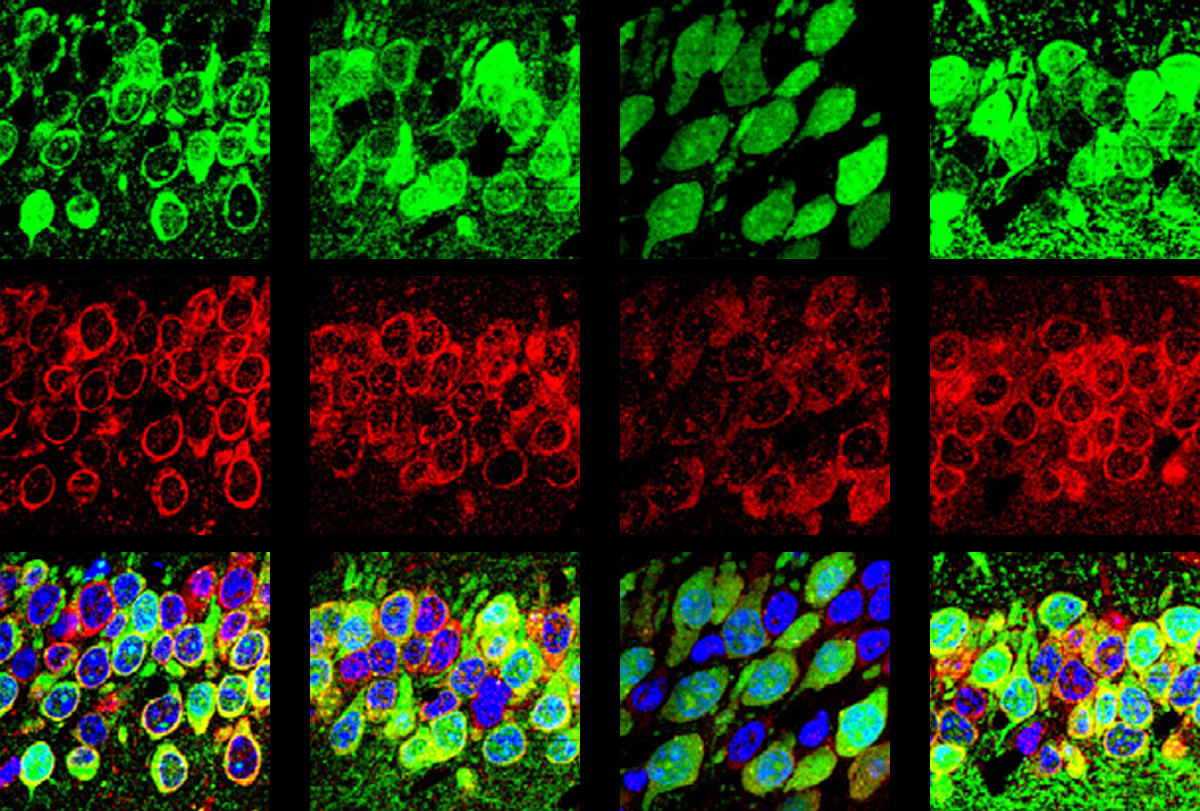
This protective pathway is suppressed in select brain cells in mice missing FMR1, the gene that codes for FMRP, the new work found. FMR1-knockout mice have reduced levels of phosphorylated eIF2-alpha (p-eIF2-alpha) and elevated protein synthesis in excitatory neurons — but not in inhibitory neurons. Postmortem brain analysis revealed decreased p-eIF2-alpha in people with fragile X.
I
The findings were published 19 July in Neuron.
In previous work by the same group, loss of p-eIF2-alpha in excitatory neurons caused the cells to become hyperactive. The results align with the signaling imbalance theory of autism, which suggests that the condition results from too much excitation or too little inhibition in the brain.
And the new study also supports previous findings that FMR1 has different effects in distinct cell types: Deleting the gene only in excitatory neurons is sufficient to trigger noise-induced seizures — a type of sensory hypersensitivity — in mice.
“I think it’s important that although FMR1 is deleted in all cell types, it has distinct effects in different cells,” Khoutorsky says. Treatments that alleviate protein synthesis in excitatory neurons may be a more effective approach than targeting all cells, he adds.