This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.

Drugs developed to treat fragile X syndrome may also work for autism because both disorders feature defects at neuronal junctions, suggests a paper published 12 January in Nature Neuroscience.
Drugs developed to treat fragile X syndrome may also work for autism because both disorders feature defects at neuronal junctions, or synapses, suggests a paper published 12 January in Nature Neuroscience1.
Researchers showed that CTEP, a drug designed to target the defect in fragile X syndrome, alleviates cognitive problems in mice that model deletion of the autism-linked 16p11.2 chromosomal region.
“I think that’s a particularly important insight, because we now know that there are hundreds of different gene mutations that probably cause autism,” says lead researcher Mark Bear, professor of neuroscience at the Massachusetts Institute of Technology. “But if those different gene mutations converge on a finite number of physiological processes, then a treatment developed for one may apply to others.”
Still, more work is needed to clarify how the drug works in the 16p11.2 model before advancing to clinical studies, experts say.
In 2002, Bear’s team discovered that mouse models of fragile X syndrome produce too many proteins at synapses2. This overproduction is mediated by mGluR5 — a receptor for the excitatory neurotransmitter glutamate — so many experimental fragile X drugs, including CTEP, block the mGluR5 receptor.
In clinical trials of fragile X syndrome, many of these candidates did not improve irritability, hyperactivity or lethargy in teenagers and adults with fragile X syndrome. The new study suggests that blocking mGluR5 may ease cognitive problems, which those trials did not assess.
“This is very exciting,” says Randi Hagerman, medical director of the MIND Institute at the University of California, Davis. Hagerman was not involved in the study, but has led clinical trials of other fragile X drugs. “I think that the mGluR5 antagonists are going to be helpful for young children with fragile X, but I think they could also be helpful for many young children with autism, particularly this 16p11.2 microdeletion subgroup,” she says.
Several autism-linked mutations affect protein synthesis at synapses. This led Bear and his colleagues to hypothesize that flawed protein synthesis underlies some forms of the disorder3.
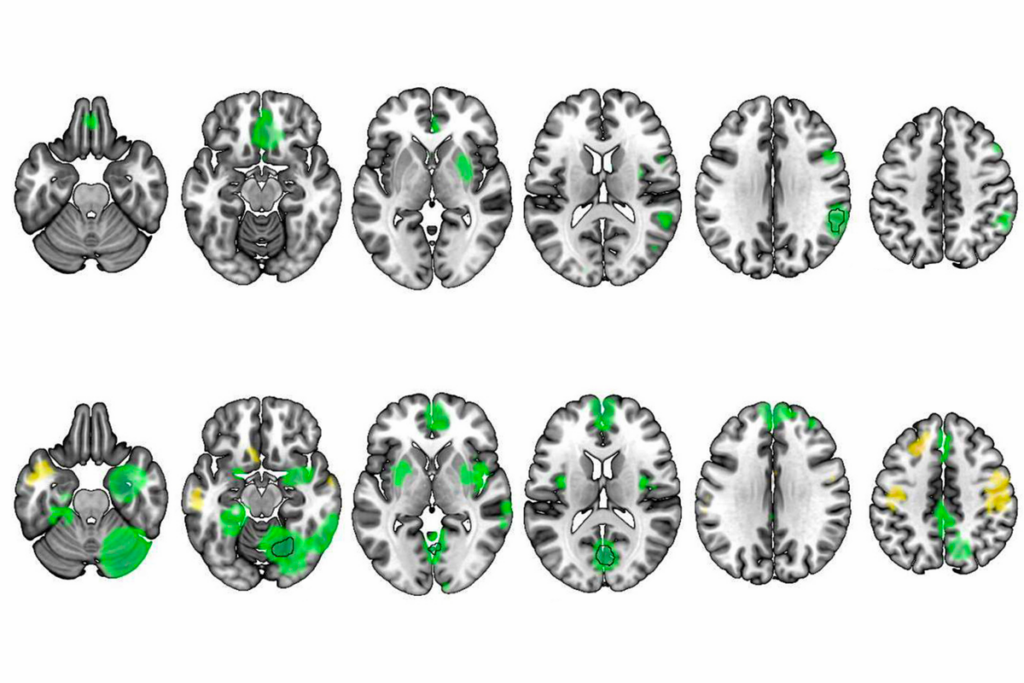
To test this theory, the researchers studied mice modeling a deletion of 16p11.2, one of the most common genetic risk factors for autism. Three genes in this chromosomal region are known to play a role in mGluR5 signaling.
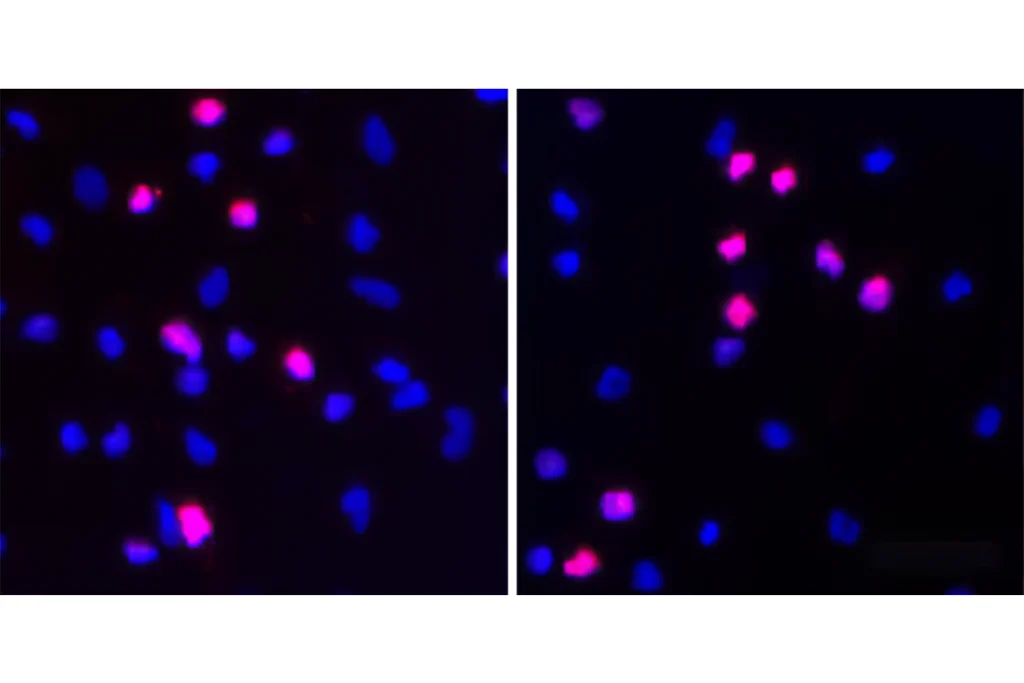
The researchers found altered protein synthesis at synapses in brain slices from the mice. The animals also have problems with learning and memory, similar to mouse models of fragile X syndrome. Treatment with CTEP every other day for four weeks alleviates this cognitive deficit in both sets of mice.
This apparent overlap may come as welcome news to drug companies developing mGluR5 inhibitors.
“I think the importance of this paper is to show that you can normalize physiology with the same class of drugs in two different diseases that cause common features clinically,” says Elizabeth Berry-Kravis, professor of pediatrics at Rush University Medical Center in Chicago. She was not involved in the study but has led trials of arbaclofen, a leading fragile X drug candidate.
“That’s good news for drug development, because to think that we’re going to develop a different drug for every single form of autism when there may be hundreds of forms of autism is unlikely,” she says.
The researchers still need to resolve one seeming contradiction: Fragile X mice make too many proteins at synapses, whereas mice modeling the 16p11.2 deletion make too few.
“It was a head-scratching moment,” Bear says. “It was exciting that once again we have a disorder that causes autism and intellectual disability that is also characterized by disrupted protein synthesis, but we didn’t expect it to be going down.”
1. Tian D. et al. Nat. Neurosci. Epub ahead of print (2015) PubMed
2. Huber K.M. et al. Proc. Natl. Acad. Sci. USA 99, 7746-7750 (2002) PubMed
3. Kelleher R.J. and M.F. Bear Cell 135, 401-406 (2008) PubMed