This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
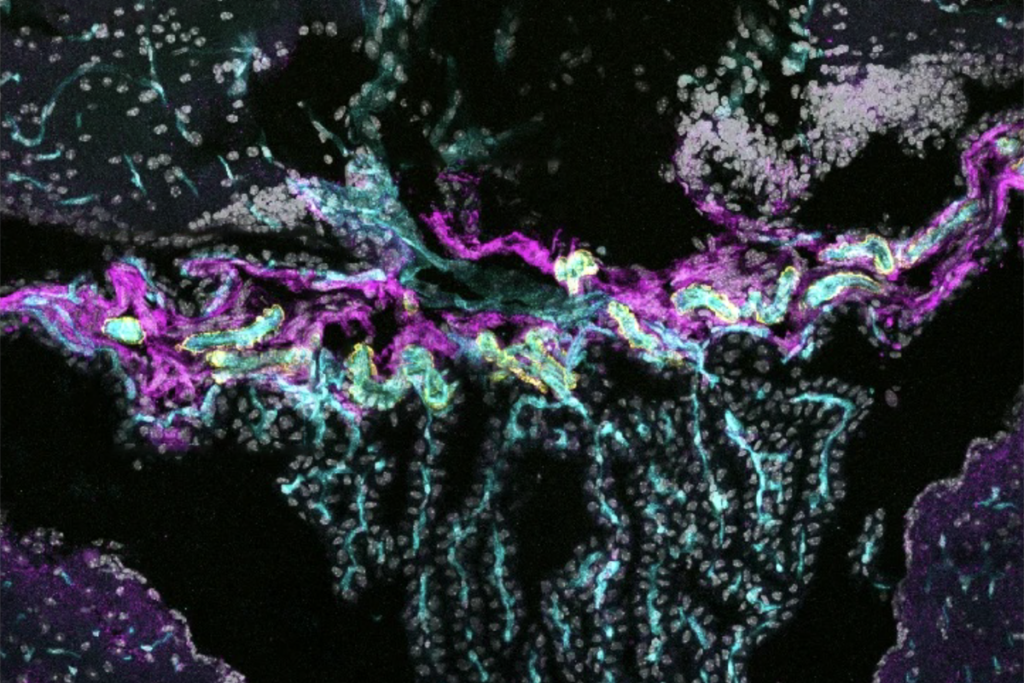
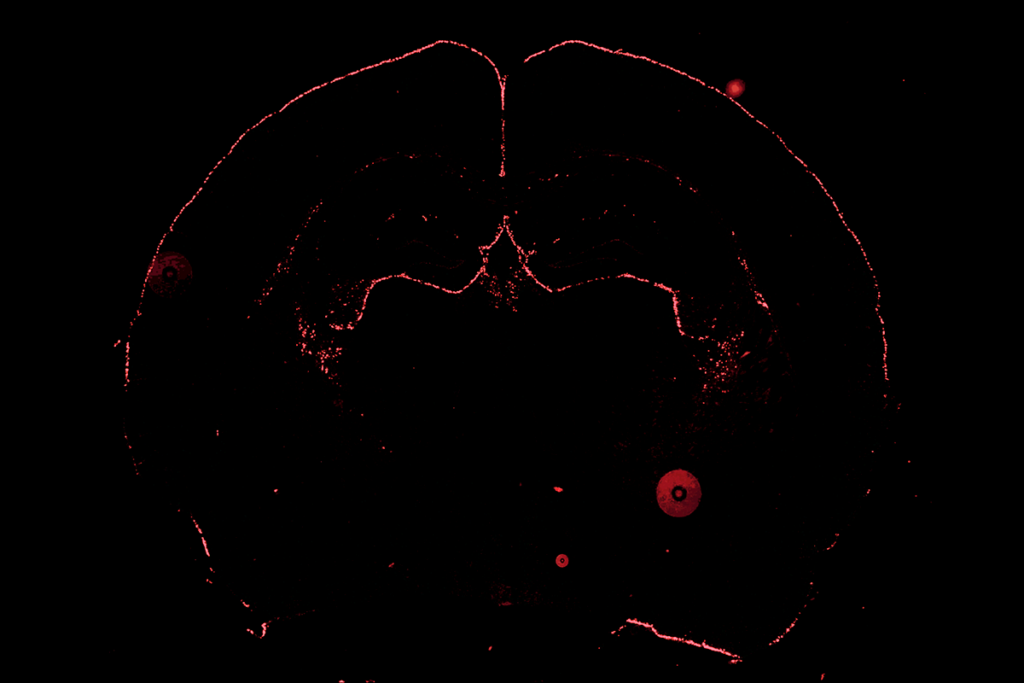
Neurons from people with Timothy syndrome, and from mouse and rat models of the disorder, have defects in the growth of their branches, according to a study published 13 January in Nature Neuroscience.
Neurons from people with Timothy syndrome, and from mouse and rat models of the disorder, have defects in the growth of dendrites — branching structures that help brain cells communicate — according to a study published 13 January in Nature Neuroscience1.
Timothy syndrome, a rare disorder affecting the nervous system and the heart, is caused by a mutation in the gene CACNA1C. The gene codes for a calcium-pumping channel in the cell membranes of neurons. People with Timothy syndrome often have features of autism.
When healthy neurons are stimulated, dendrites tend to grow longer and form branches — though dendrites are constantly growing and then retracting, the dominant force is toward greater length. Researchers have linked impairments in the growth of dendrites with autism and other neurodevelopmental disorders2.
The researchers found that when they stimulated rat neurons bearing a mutation in CACNA1C, the dendrite arbors shrank in length and complexity, as retraction events outnumbered growth spurts.
They saw a similar effect in neurons grown from induced pluripotent stem cells from individuals with Timothy syndrome. Mice that carry the CACNA1C mutation also have shorter dendrites than controls do.
Calcium channels have shown up in genetic screens for autism before, leading researchers to wonder whether too much calcium in neurons may underlie the disorder’s neurodevelopmental problems.
At least in this case, however, the dendrites are unaffected by the amount of calcium the channel pumps into the neurons, the researchers found. The growth defect stems instead from a cascade of cellular signals set in motion when the channel binds to another protein in the cell.
The mutant version of the channel is less effective at binding to a protein called GEM, the team found. When GEM binds to the channel, a protein called RhoA, which is known to contribute to dendrite retraction, is inactivated — although the precise mechanism is still unclear. Neurons with the mutant version of the channel have excess RhoA, the study found.
1. Krey J.F. et al. Nat. Neurosci. 16, 201-209 (2013) PubMed
2. Kaufmann W.E. et al. Cereb. Cortex 10, 981-991 (2000) PubMed