This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
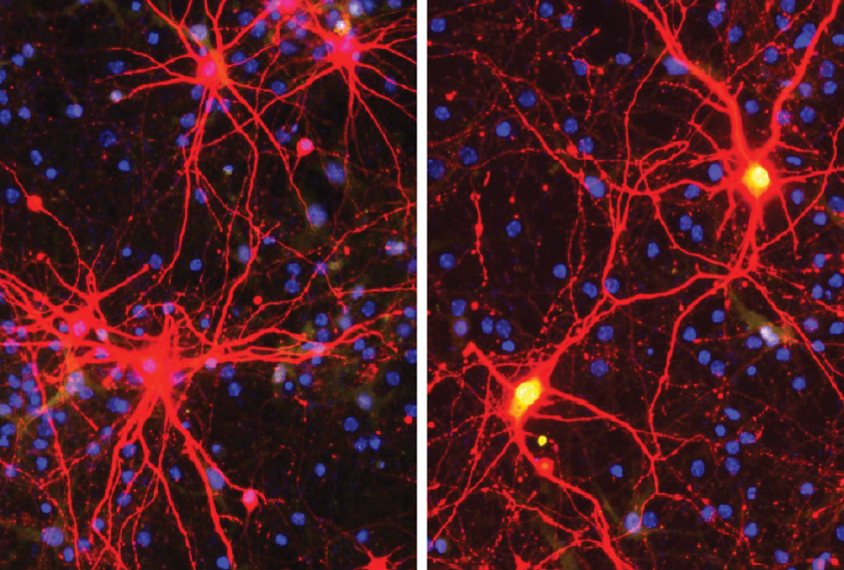
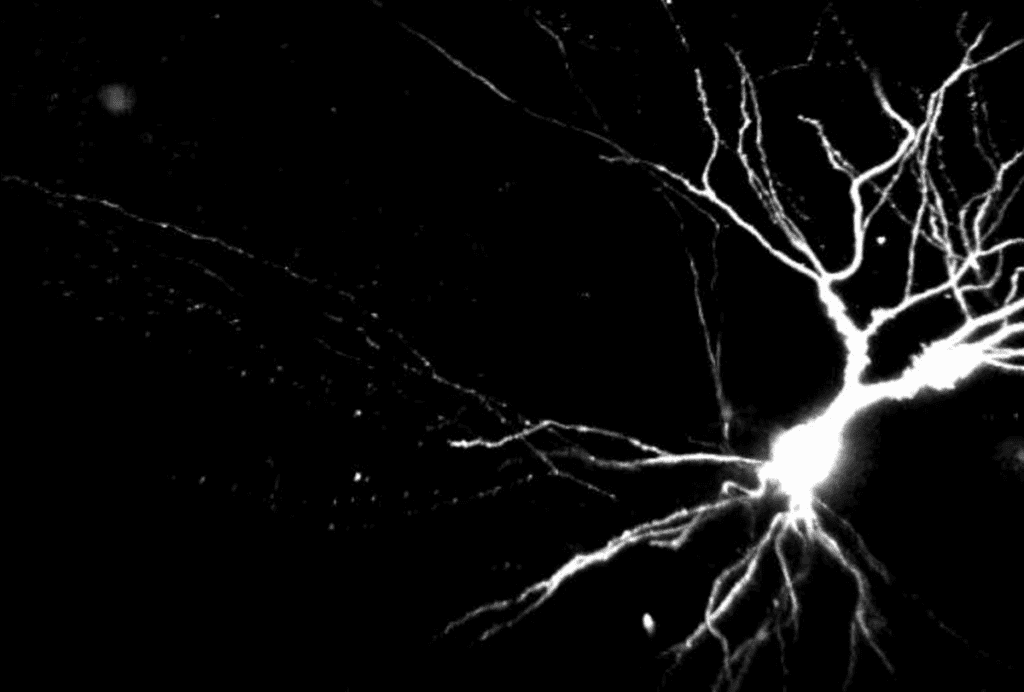
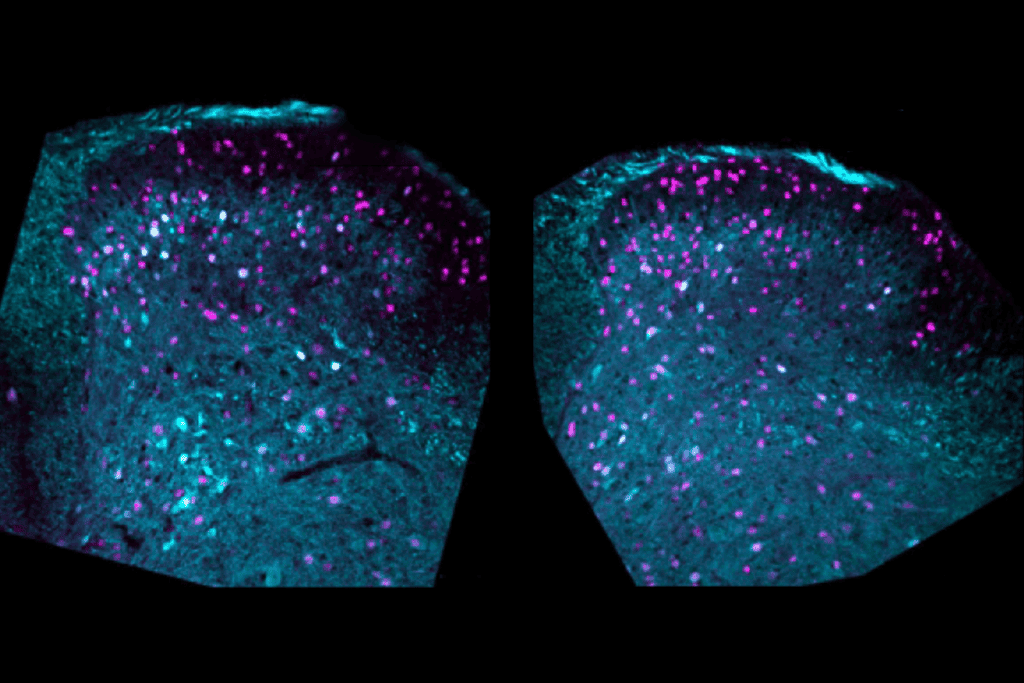
A gene-editing treatment shows long-lasting effects in a mouse model of Angelman syndrome, a genetic condition related to autism.
Editing DNA in embryonic and newborn mice by using CRISPR technology can override mutations underlying Angelman syndrome and prevent many of the condition’s traits, according to a new study1. The effects last for at least 17 months and may be permanent, the researchers say.
“It’s very exciting,” says Steven Kushner, professor of psychiatry at Columbia University, who was not involved in the study.
Angelman syndrome usually stems from a mutation in or deletion of the UBE3A gene. People have two copies of the gene — one from each parent — but typically only the one passed down from the mother is active in neurons. Mutations that stymie that copy can lead to a lack of UBE3A protein in the brain, causing the syndrome’s core traits: developmental delays, motor dysfunction, speech impairments, seizures and, often, autism.
These traits improve in response to treatments that activate the silent yet intact paternal copy of UBE3A and boost production of the protein in Angelman syndrome model mice2,3. But these treatments wear off over time, requiring repeated injections into the spinal fluid or brain.
The new therapy is effective after only two doses, says lead researcher Mark Zylka, professor of cell biology and physiology at the University of North Carolina at Chapel Hill.
The strategy uses the enzyme CRISPR-Cas9 to cut and edit DNA encoding an ‘antisense RNA’ molecule that ordinarily serves to block production of UBE3A protein from the paternal copy of the gene. The technique also rouses the silent paternal copy of the gene in cultured human neurons, suggesting that it might work in people.
Treated mice did not show any negative side effects or unintended mutations. But other researchers warn the approach may still have rare ‘off-target’ effects that could lead to birth defects or cancer.
“That’s one of the big reasons why the use of CRISPR in humans is still something that is being taken very, very cautiously,” Kushner says.
Zylka and his colleagues used a modified virus to carry the gene-editing enzyme into mouse cortical neurons. They tested 260 different ‘guide RNAs’ to find one that could escort the enzyme to the intended region of the genome without damaging other genes.
The team injected the therapy into the brains of Angelman mice — first when the mice were in the womb and again when they were a day old.
The double dose delivered the treatment to all layers of developing cortex, the researchers found. And it activated the paternal copy of UBE3A in every brain region they checked, aside from the cerebellum, until the mice were 17 months old. The effects likely last even longer, they say.
Mounting data show that a virus like the one they used can insert itself into the genome, resulting in permanent changes to an animal’s DNA, Zylka says. When that happens, the viral genome can affect nearby genes.
In this case, the viral genome contains instructions to tack extra nucleotides — the building blocks of the genetic code — on to messenger RNA. These additions render ineffective the antisense RNA that normally silences paternal UBE3A.
The injections prevented behaviors typically seen in Angelman mice, the researchers reported in Nature in October. Treated mice showed less hind-limb clasping, considered akin to repetitive behaviors seen in autistic people. They also spent more time in the center of an open field, suggesting they are less anxious than untreated mice, and performed better on a test of motor coordination.
Mice that model Angelman syndrome tend to have smaller brains than typical mice, but this trait, too, was at least partially averted in the treated animals.
The approach did not block all Angelman traits, however: The mice still engaged in marble-burying, another repetitive behavior; and only female mice showed improvements in obesity.
The treatment may not have worked in enough cells to correct all traits: The researchers found UBE3A protein expressed in 58 percent of the model animal’s cortical neurons, whereas typical mice have it in all neurons.
It is also possible that different traits have unique critical windows for treatment, says Stormy Chamberlain, associate professor of genetics and genome sciences at the University of Connecticut in Farmington, who was not involved in the study. Studies have shown that the earlier UBE3A is reinstated, the more Angelman traits are ameliorated.
Further research may improve the treatment’s effectiveness in mice, but its utility for people could be limited by its reliance on active CRISPR enzyme, which can introduce unpredictable cuts and mutations in DNA, Chamberlain says.
Even so, “it’s important to keep an open mind and try some of these ‘the sky’s the limit’ approaches,” she says.
Other gene-based therapies for Angelman syndrome also pose safety concerns. An ongoing trial of a different treatment, which activates paternal UBE3A using strands of modified RNA, had to be halted because it causes temporary leg weakness in children with Angelman syndrome. Researchers hope to resume that trial with a different dosing plan.
Zylka and his colleagues plan to try their approach with an alternate version of CRISPR — one that cannot cut DNA but can still activate paternal UBE3A — which may be safer for use in people.