Listen to this story:
0:00
/
Adjusting genetic analyses could help plug autism’s heritability gap, according to a new preprint.
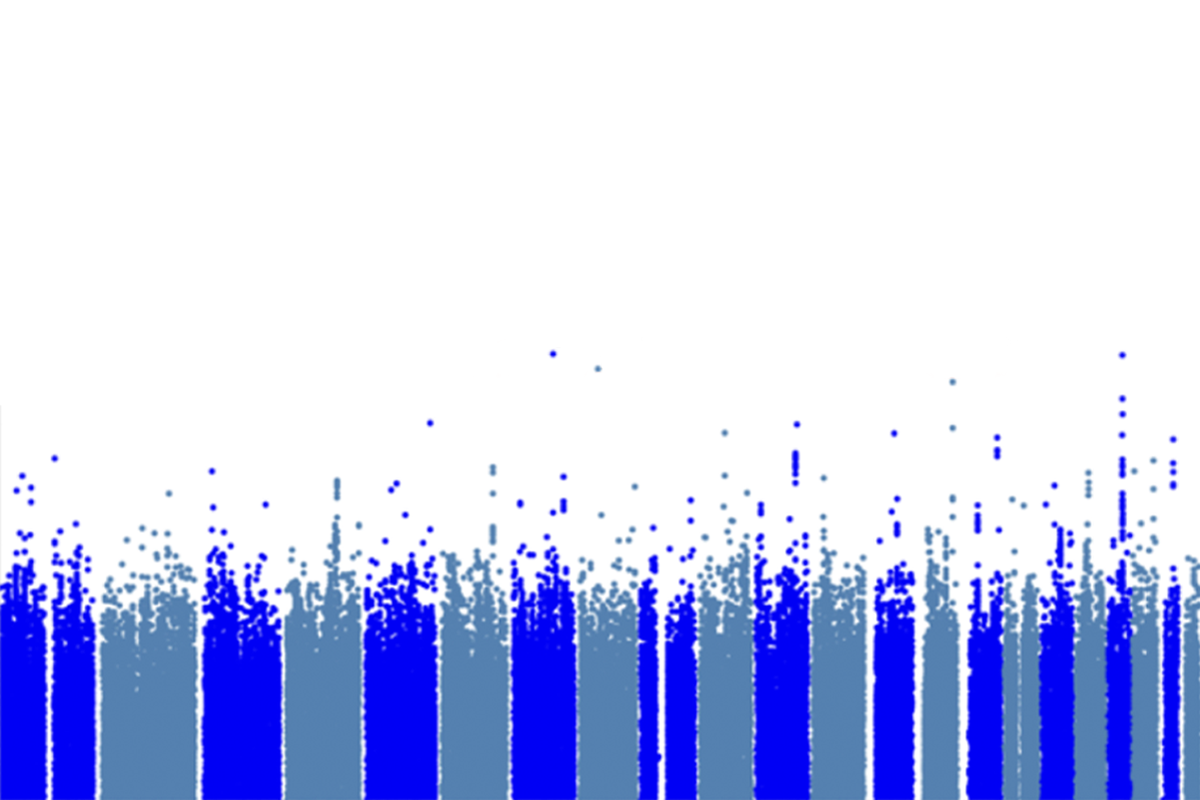
The stringent statistical threshold used in genome-wide association studies (GWAS) hides low-frequency variants linked to autism and other conditions, a new preprint suggests. Lowering the threshold could reveal some of those variants, according to the study.
“It’s really innovative,” says Carrie Bearden, professor of psychiatry and biobehavioral sciences at the University of California, Los Angeles, who was not involved in the work. Provided the findings are replicated, a revised threshold could capture “a lot of the potential signal that we’re missing” for autism’s heritability, she says.
Most known genetic changes linked to autism consist of common variants, those found in more than 5 percent of the population, and rare variants, which occur in less than 1 percent of people. But few studies have uncovered variants that sit between those extremes.
Such low-frequency variants may account for some unexplained heritability but are concealed by standard statistical parameters, says study investigator Marla Mendes, a research associate at the SickKids Research Institute in Toronto.
The conventional GWAS threshold of a p-value of less than 5 x 10-8 is tailored to searches for common variants; when a genetic change is shared by a smaller population, there is less statistical power, and real associations can be missed, says study investigator Elemi Breetvelt, adjunct scientist and principal investigator at the SickKids Research Institute. “Maybe we are missing meaningful information by applying methods that were developed for common traits like height or BMI.”
Mendes, Breetvelt and their colleagues generated an alternative threshold by applying the Bonferroni correction—a statistical method that minimizes false positives during analyses that involve multiple statistical tests. By accounting for linkage disequilibrium—the fact that two nearby loci are likely to be inherited together—the team could identify the number of independent tests and drop the significance threshold.
They then varied that threshold and simulated its ability to detect known autism variants. That test run pointed to 2.03 x 10-7 as the optimum limit for detecting low-frequency variants: That threshold identified the most real variants without increasing the rate of false positives within the simulation.
U
The study “challenges a long-standing assumption in statistical genetics—that a single genome-wide significance threshold works equally well across all allele frequencies,” says Aaron Besterman, medical director at the Laura Rodriguez Research Institute of Family Health Centers in San Diego, who was not involved in the study. “Raising that question is valuable for the field.”
As large-scale genetic databases become more accessible, “we may need more nuanced statistical approaches to capture the full complexity of autism genetics,” says Maria Chahrour, associate professor of neuroscience and psychiatry at the University of Texas Southwestern Medical Center, who was not involved in the work.
“But the ultimate test” is whether it can be replicated using an independent dataset, Chahrour says. Until then, we won’t know whether the proposed threshold can generate meaningful associations, she adds.
And although the simulation didn’t produce false positives, boosting statistical power is bound to yield some inaccurate hits in a larger dataset, says Michael Salter-Townshend, associate professor of statistics at University College Dublin, who was not involved in the work. “There’s no such thing as a free lunch,” he says.
The preprint investigators plan to repeat their analysis in separate datasets, including data derived from non-European populations, Mendes says. Rare and common variants vary more in people of African ancestry than in those of European descent, so it’s likely that such populations also carry distinct low-frequency variants, she adds.
Mendes and her colleagues hope to generate some of those data themselves, allowing them to optimize their search for less-common variants, she says. For instance, typical GWAS databases contain imputations—inferences that replace absent genetic data—that tend to be less accurate for low-frequency variants.
By collecting whole-genome sequences from people with autism, the group can create complete genetic databases that are geared toward uncommon variants. “Our hope is not only to replicate the signals we observed but also potentially identify additional loci,” Mendes says.