Listen to this story:
0:00
/
The approach prompts cultured cells to correct the genetic mutation in fragile X syndrome using their own DNA repair system, but it still needs to be tested further.
Hun-Goo Lee did not set out to discover a new way to potentially treat fragile X syndrome. He just wanted to solve a mystery: Why do some cells with a fragile X mutation remain unaffected?
The mutation — more than 200 copies of the trinucleotide string ‘CGG’ in the FMR1 gene — typically silences the gene’s expression and prevents production of the protein FMRP. Under some lab conditions, though, embryonic cells that carry these long CGG repeats still produce FMRP.
That FMRP production arises because the culture conditions enable the cells’ DNA repair machinery to spot the CGG repeats and remove them, according to a new study from Lee and his colleagues. Inspired by this mechanism, the team developed a novel gene-editing tool that enables other types of fragile X cells to remove the trinucleotide repeats too.
“This is exciting work,” says Antonio Bedalov, professor in the division of translational sciences and therapeutics at Fred Hutchinson Cancer Center in Seattle, Washington, who was not involved in the study. This approach could, in theory, treat fragile X syndrome and other conditions with similar genetic repeats, he says. “You have really attacked the root cause of the problem.”
E
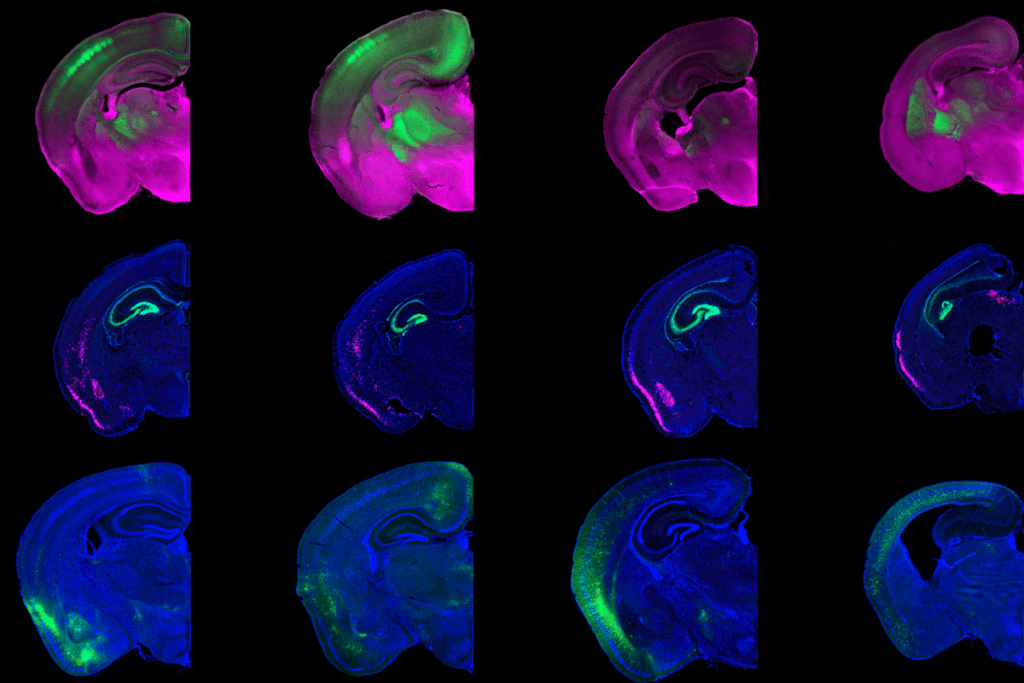
mbryonic fragile X cells can appear unaffected and express FMR1 when cultured in a specific media, and two drugs in that media were responsible, Lee’s team found.In fragile X syndrome, the CGG repeats in FMR1 accumulate epigenetic tags called methyl groups, which silence the gene. But these two drugs induce demethylating enzymes, which removes the methyl tags and restores FMR1 expression in stem cells and neural progenitors derived from people with fragile X, they discovered. The treatment led to the excision of the CGG repeats. The contraction began after three to six days of treatment; by 12 days, it was complete.
The researchers got similar results when they used an existing version of CRISPR to target a methyl-removing enzyme to the FMR1 gene.
But delivering an inactive version of the enzyme as a control also accomplished the same thing, suggesting that demethylation was not the only driving factor for CGG contraction to occur. “This was the most mind-boggling thing,” says lead investigator Jeannie Lee, professor of genetics at Harvard Medical School and acting chair of the department of molecular biology at Massachusetts General Hospital. Hun-Goo Lee is a research fellow in Jeannie Lee’s lab at Harvard.
The researchers then realized that the CRISPR-based approach and the demethylation process can both cause DNA to form a structure called an R-loop. These loops occur when DNA’s double strand becomes interrupted by a segment of single-stranded RNA — akin to a bit of fabric getting caught across a strip of Velcro and creating a bump where the adhesive strips can no longer connect.
This genomic ‘bump’ — stabilized by the large number of CGG repeats in the fragile X cells — activates a cell’s DNA repair machinery, which removes repeats.
The team then designed a catalytically inactive version of CRISPR that specifically prompts CGG repeats to form R-loops. The tool could eventually be packaged in viruses and delivered to the brain, the researchers say. The results were published in May in the journal Cell.
“I like the idea of using the endogenous repair pathways because they come with millions of years of evolution” to make things happen with the highest fidelity, Jeannie Lee says.
A
lthough the work is preliminary, it “gives a ray of hope into the fragile X field,” says Randi Hagerman, distinguished professor of pediatrics and medical director of the MIND Institute at the University of California, Davis, who was not involved in the study. “It means you could eliminate methylation and then cut down on the CGG repeats without having to use gene therapy intervention.”But whether that treatment would come with any repercussions in people is unclear, Hagerman adds. For one, the method removes enough CGG repeats to put a cell below the 200-copy mark that silences FMR1, but not necessarily enough to put it in the typical range of less than 55. People with 55 to 200 repeats have a fragile X “premutation” — one that may not result in intellectual disability but can cause other symptoms early in life, including fragile X–associated neuropsychiatric disorder, anxiety and depression. “These concerns about premutation involvement must be considered before doing a treatment that changes a full mutation to a premutation,” Hagerman says.
The approach “is not without a risk,” Bedalov agrees. Still, he says, there is likely a benefit in using the catalytically inactive version of CRISPR, which “is not going to be making cleavages all over the genome.” And the tool is smaller than typical for CRISPR-based approaches, making delivery to the brain simpler, he adds.
Before any conclusions are made, the findings need to be tested in non-dividing cells, Bedalov says. DNA repair is active during cell division, he notes, and thus would readily occur in the stem cells and neural progenitor cells used in the study. “Given that neurons do not divide, it will be important to demonstrate that R-loops can trigger repeat excision in the non-dividing cells,” he says.
The researchers plan to test that next. Ultimately, Jeannie Lee says, the work could lead to a way to remove excess trinucleotides in people, which could help treat fragile X syndrome as well as other conditions that stem from this form of mutation.
“We started here,” she says, pointing to the ground, referring to the initial puzzle she and Hun-Goo Lee set out to solve. “And somehow we ended up in outer space.”