This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Integrating human-specific genetic elements into mice may provide a permissive, ‘humanized’ environment for studying autism.

Genetic studies over the past several years have identified dozens of genes that carry mutations associated with risk for autism. These discoveries promise to help us understand the developmental origins of autism in the brain.
However, we cannot study the effects of mutations directly in people. We instead use animals as living models that recapitulate features of the developing human brain, and that we can engineer and manipulate in order to understand those features.
The more closely an experimental model reflects human biology, the more relevant its results will be to people’s well-being. But to fully understand autism, we must manipulate animal models to take into account aspects of the human brain that are uniquely human.
Researchers commonly use mice for modeling human brain development and the conditions associated with it. The human and mouse brain develop in generally similar ways, and it is technically straightforward to design and generate mutant mice.
The autism risk genes discovered to date are especially amenable to study in mouse models. Mice have analogous versions of these genes, and the autism-linked mutations within them damage or destroy protein function. Researchers can therefore model the functional effects of human mutations by disrupting the corresponding mouse gene.
However, mouse models have one clear limitation: They are not human. This may seem to be an obvious statement, and yet because we rely so heavily on mice to understand human genetic conditions, it is often easy to forget.
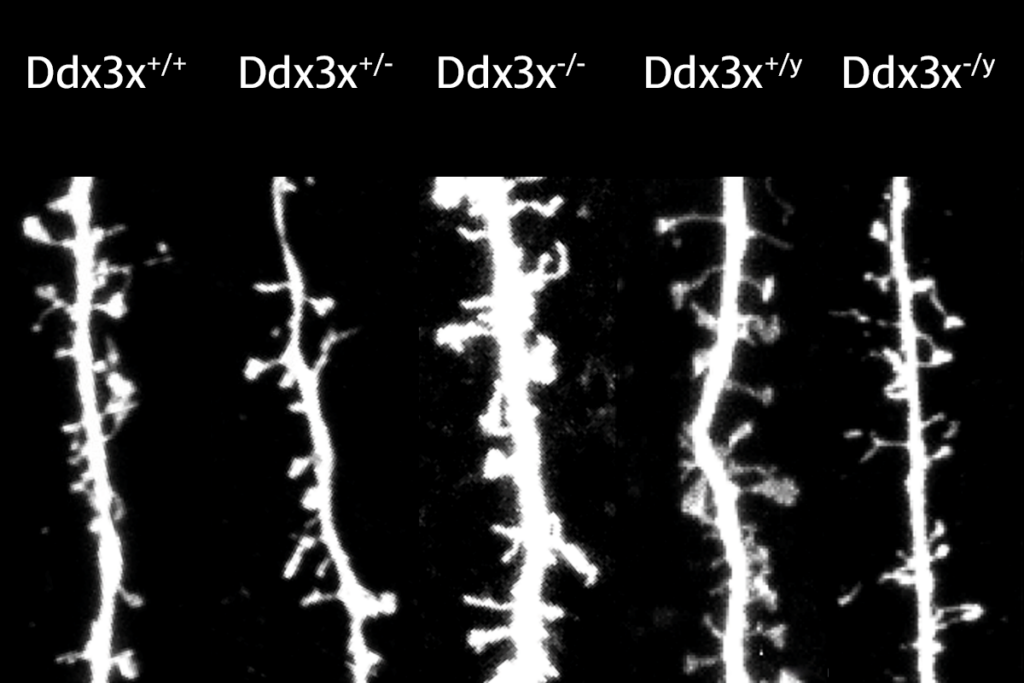
For example, one prominent autism risk gene, CHD8, has been studied using at least four strains of mice missing the gene. One of the primary functions of CHD8 is to control the expression of other genes during brain development, and it appears to control many of the same genes in people and mice1,2. There is little doubt that these studies are providing novel and important insights into how loss of the CHD8 protein alters molecular and developmental processes in the mammalian brain.
And yet, loss of CHD8 function in a person may result in changes in brain development and circuits — and, ultimately, behavior — that cannot be modeled in mice.
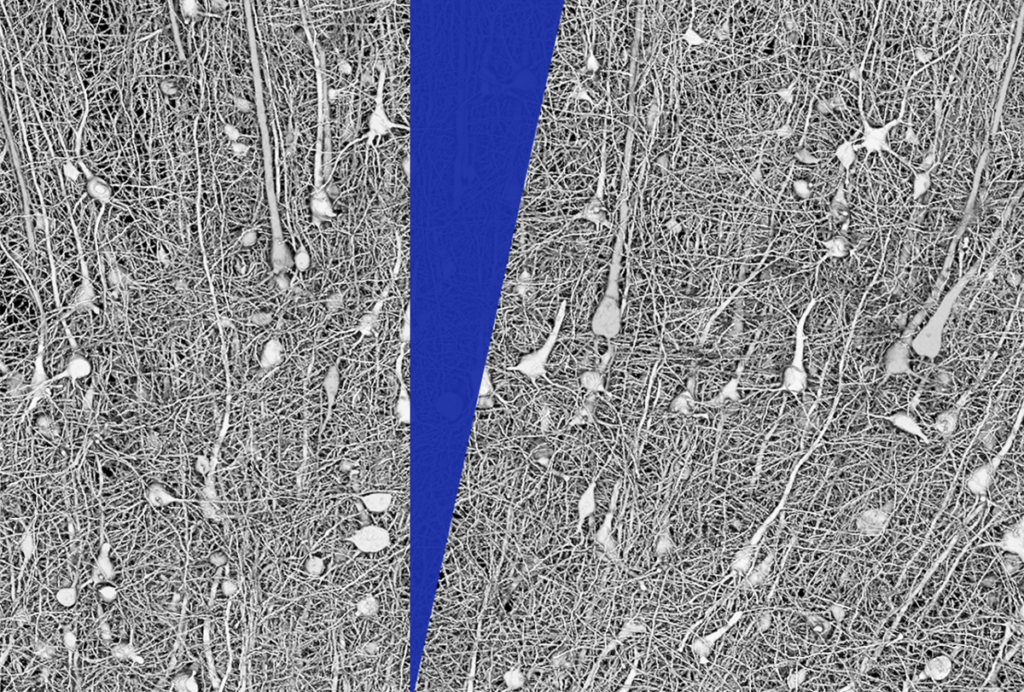
The unique expansion and elaboration of the human brain — in particular, the cerebral cortex —distinguishes us from other species, including the mouse. The significance of these characteristics is only now beginning to be understood.
For instance, autism involves aspects of human social cognition — how we understand and interact with others around us — that may be problematic to model in mice. Social cognition in people is likely to require functional processes that are unique to the human brain. Such processes may well be disrupted in autism; however, they may not be represented in the existing mouse models of the condition.
How can we make the mouse a better model of human brain development?
We can start with emerging research over the past decade: Multiple studies have identified uniquely human genetic changes that may have contributed to the evolution of the human brain.
In comparisons of genomic sequences across many species, two distinct classes of elements stand out in the human genome: human accelerated regions, or HARs, and human-specific gene duplications, or HSGDs3,4.
HARs are non-gene-coding sequences that are highly conserved across many species, but which are surprisingly different in people. Many of these regions encode ‘regulatory switches’ with human-specific functions, leading to uniquely human differences in when, where and at what level genes are expressed in the brain. Many HSGDs are expressed in the brain, where they might have particularly large biological effects.
At the same time, functional studies of the developing human brain are revealing uniquely human patterns of gene expression and regulation. The scope and resolution of these studies are increasing: There are several major publicly funded initiatives to comprehensively map gene expression and regulation in the developing human brain, and to compare human maps with those of nonhuman species5.
How might these discoveries be used to enhance the insights gained from mouse models of autism risk genes?
HARs and HSGDs themselves are prime candidates for modeling in the mouse, with the goal of discovering how they may have contributed to ancestral features of brain development and function.
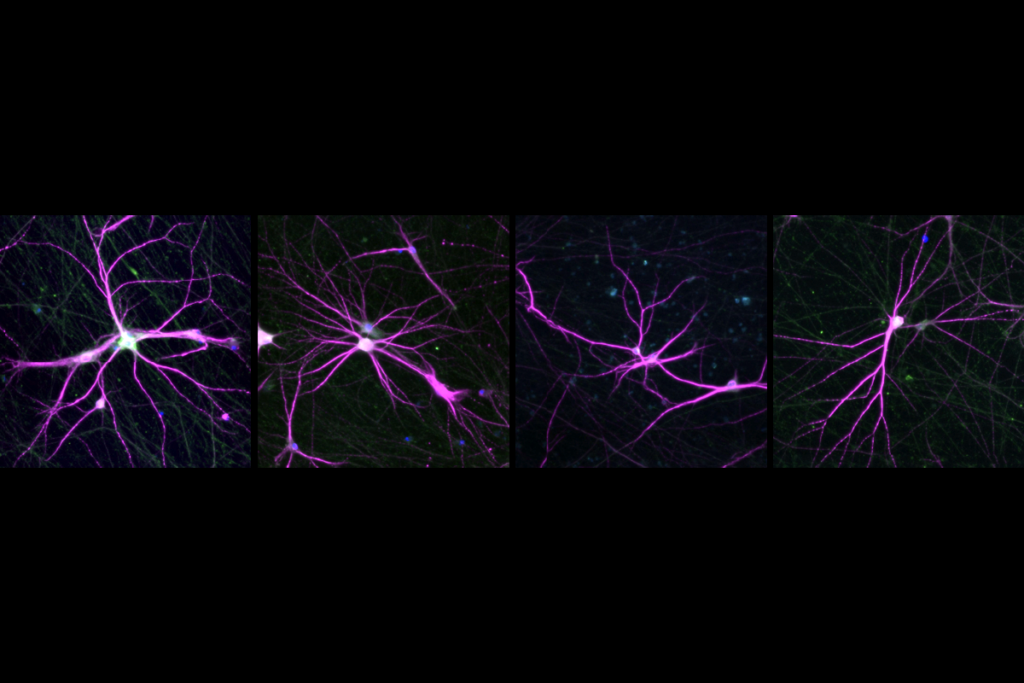
Our collaborators used this approach to study the role of one HSGD, SRGAP2C, in the developing brain6. Mice engineered to carry a copy of this gene show changes in the development and density of neuronal junctions, or synapses, that are reminiscent of circuits in the human cortex.
Mouse models of HARs and HSGDs may thus provide a permissive, ‘humanized’ environment for studying the effects of autism-linked mutations on the developing brain.
For example, deleting CHD8 in a SRGAP2C-humanized mouse may reveal effects of the gene’s loss on cortical development and circuits that are not visible in a typical mouse. Comparative studies of developing human and nonhuman brains would complement these genetic studies.
The prospect of two steadily advancing fields of scientific discovery — defining genetic mechanisms that are uniquely human, and identifying those that are associated with autism — cross-pollinating in such a way is tantalizing. By combining these bodies of knowledge, we may come to better understand the uniquely human complexities of autism.
James Noonan is associate professor of genetics at Yale University.