Update
One of the studies described in this story has since been published in Cell Genomics.
Strips that are thousands of base pairs in length offer better resolution of structural variants and tandem repeats, according to two independent preprints.
Long-read sequencing reveals novel autism-linked variants that short-read sequencing misses, according to two preprints posted on medRxiv in July.
“These are two very nice papers from top labs. They both make compelling cases for long-read sequencing,” says Michael Gandal, associate professor of psychiatry, genetics and pediatrics at the University of Pennsylvania Perelman School of Medicine, who did not contribute to either investigation. “It’s likely this will become the new gold standard.”
Conventional short-read sequencing typically outputs DNA fragments of about 150 base pairs in length. Computational tools then stitch these bits together to cover longer stretches of the genome, but they can struggle to assemble complex or repetitive regions. Newer long-read sequencing approaches produce strips that are thousands of base pairs in length, offering better resolution of structural variants (SVs) and tandem repeats, although at a higher cost.
Large-scale genomic sequencing studies have uncovered hundreds of autism-linked genes. But a large proportion of autism heritability remains unexplained. Some of this variation may come from structural variants that short reads can’t detect reliably, says Jonathan Sebat, professor of psychiatry and cellular and molecular medicine at the University of California, San Diego and an investigator of one of the studies.
“We can find new types of mutations that we just can’t see with short reads,” he says.
Sebat and his colleagues aligned long reads to a reference genome to piece together the whole genomes of 243 people from 63 families that have at least one child with autism. Comparing these long-read data with conventional short-read data for the same participants, they pinpointed 15 autism-related rare de novo SVs in coding regions, 3 of which were detectable only with long reads, the study shows.
Rather than aligning sequencing data to a reference genome, the team behind the other study used long reads to piece together near-complete, bespoke genome assemblies for 189 people from 51 families that have at least one child with autism that has no known origin. The team pinpointed three SVs that affect known autism-linked genes, as well as nine other SVs with potential functional consequences, most of which were missed by short reads.
Both studies report that long-read sequencing increased the number of autism-linked variants discovered by 4 to 6 percent.
That “might sound modest,” wrote Evan Eichler, professor of genome sciences at the University of Washington and an investigator of one of the studies, in an email to The Transmitter. But if rare gene variants act synergistically to increase autism likelihood, it will be essential to find them all. “Long-read sequencing gets us there, short-read sequencing does not,” he wrote.
S
ebat’s team pinned down 44,647 non-tandem repeat SVs using long reads, 16,488 of which had been missed by a prior short-read analysis. But roughly 7,000 variants were detectable only by short reads, highlighting the value of using both technologies, Sebat says.Most SVs occurred in noncoding regions of the genome, the study shows. But the 15 de novo SVs—uninherited variants that are clearly linked to a child’s condition—were located in functional regions in the exome. Of these, three were identified only with long reads, six only with short reads and six with both methods.
“In some cases, long-read sequencing was finding variants in the exome,” Gandal says. “It’s significant because that in theory shouldn’t happen.” Short reads, he says, were thought to be accurate enough to resolve the 1 to 2 percent of the total genome that codes for genes. This shows that “that isn’t the case,” he adds.
One of these de novo SVs resulted from an in-frame duplication of STK33, which caused a “radical” expansion of the protein product’s helical domain, as predicted by a protein folding analysis—indicating that the variant likely has functional consequences, Sebat says. Two other variants, one de novo and the other inherited, resulted from complex rearrangements caused by a duplication event following a deletion, further sequence-level characterization revealed.
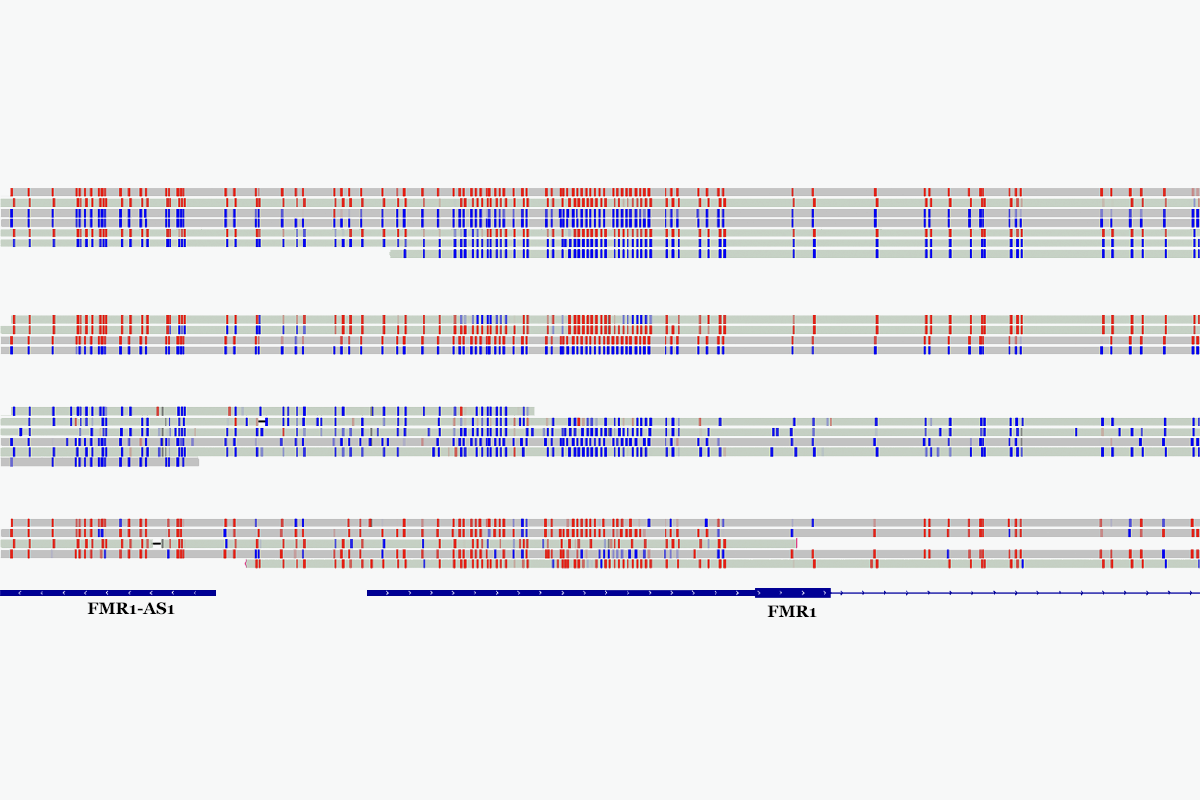
Long-read sequencing also enabled chromosome phasing—the process of piecing together alleles on each parental chromosome—and methylation analysis. Phasing the X chromosomes in 43 female participants revealed that the trinucleotide repeat length of FMR1, which is associated with fragile X syndrome, correlates with the gene’s methylation levels but is independent of X chromosome inactivation, a finding that aligns with previous studies. “This makes the case for using both short reads and long reads,” Gandal says.
I
n the other study, Eichler and his colleagues compared each participant’s genome with the human pangenome reference sequence and data from the Human Genome Structural Variation Consortium, which enabled them to whittle their initial set of 33,548 SVs down to 46 total SVs of interest in the 87 children in their cohort.Three of these de novo SVs—two of which were absent in a comparable short-read analysis—overlapped with known autism-linked genes. And nine de novo SVs coincided with exonic or regulatory regions, seven of which were missed by short reads.
Chromosomal phasing with these data revealed six people who each carry a rare autism-linked SV in both copies of a gene. None of the six variants identified through this analysis are found in the pangenome or in a control cohort of more than 14,000 people.
“I’m somewhat surprised by how frequently observed they are,” given that the odds of a homozygous variant occurring on both parental chromosomes are low, says Charleston Chiang, associate professor of population and public health sciences and quantitative and computational biology at the University of Southern California Keck School of Medicine.
These homozygous SVs were not as evident in the other study. But this may be because the pangenome study focused exclusively on autism cases with no known origin, mainly in affected females, Gandal says.
Because of the high cost of long-read sequencing, both studies are limited by the scale at which the technology can be deployed, Chiang says. As long-read sequencing becomes more affordable and can be applied to larger groups of people, it will likely become ubiquitous, he adds.
Gandal agrees: “This is just the tip of the iceberg.”