Dialing up the activity of certain receptors in the amygdala reverses some traits of fragile X syndrome in a rat model of the condition, according to a new study.
The finding contradicts the prevailing idea that overactive signaling through these receptors underlies the syndrome’s traits — and offers new clues as to why potential fragile X drugs have failed in clinical trials.
“It’s a really important paper,” says Randi Hagerman, medical director of the MIND Institute at the University of California, Davis, who was not involved in the work. “I think that we need to translate these kinds of research findings as soon as possible to human clinical trials.”
Fragile X syndrome is the most common form of inherited intellectual disability and is often accompanied by anxiety, seizures and autism. It typically stems from mutations that silence a gene called FMR1, hampering production of the protein it encodes, FMRP.
According to a long-held theory, the dearth of FMRP overactivates a receptor called mGluR5, which in turn impairs neurons’ ability to adjust the strength of their synaptic connections. The idea is based largely on findings in model mice: Compared with controls, their synaptic links more readily weaken among cells that fire out of sync in the hippocampus, a brain area important for learning and memory. Drugs that block mGluR5 appeared to normalize the synaptic weakening and reverse traits in the mice, but those results ultimately failed to translate to people.
“One size does not fit all,” says the new work’s lead researcher, Sumantra Chattarji, senior professor of neurobiology at the National Center for Biological Sciences in Bangalore, India.
Opposite effects:
The trial failures made sense, Chattarji says, because mGluR5 does not function the same way throughout the brain. In the amygdala — an area that shapes fear and anxiety — mGluR5 mediates the strengthening, rather than the weakening, of synapses, according previous research. Fragile X mice have typical levels of synaptic weakening in the amygdala, Chattarji and his colleagues found in 2010, but they have an impaired ability to strengthen connections among synchronous neurons. And blocking mGluR5 there did not reverse the deficit.
“It bugged me,” Chattarji says. “If the plasticity is opposite, then we have a problem” when it comes to treatment.
So for the new work, Chattarji and his colleagues revisited synaptic plasticity in the amygdala using rats that lack one copy of FMR1. Rats have a larger amygdala than do mice do, making the experiments easier.
They trained the rats to associate a tone with a mild electric shock to the foot. Unlike wildtype rats, fragile X rats do not freeze out of fear upon hearing the same tone a day later, the team found, suggesting that the animals’ fear memory is impaired. Fragile X rats also had an atypical neuronal response to the tone, according to recorded electrical signals from the animals’ amygdala.
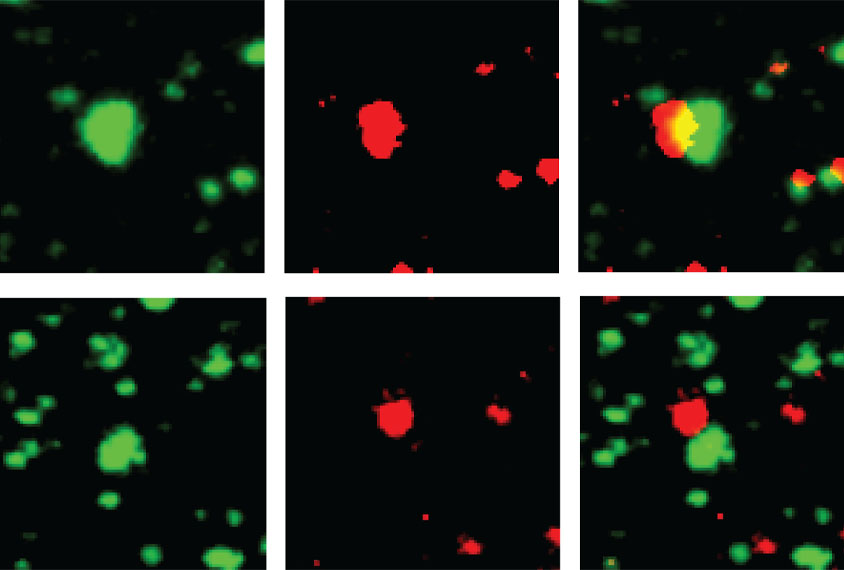
Electrically stimulating two neurons in close succession strengthened the synapses in slices of brain tissue from the amygdalae of wildtype rats — a type of plasticity that can also be induced in the hippocampi of fragile X mice, according to previous research. But in tissue slices from the amygdalae of fragile X rats, the plasticity failed, replicating the mouse findings from the 2010 paper.
The reason for the different responses in the amygdala and hippocampus was initially unclear, Chattarji says. But one clue was that, in the amygdala, mGluR5 is mostly located at the signal-sending ends of neurons, whereas in the hippocampus, it is mainly present at the signal-receiving ends, the researchers found.
Activating mGluR5 in slices of brain tissue from the amygdala of fragile X model rats reversed the deficits in synaptic plasticity, the new study showed. And infusing an mGluR5-activating drug into the amygdala of fragile X rats restored the animals’ fear recall and resulted in typical neuronal responses to the tone. The findings were published in October in Cell Reports.
Complex targets:
“If it’s mainly the amygdala that’s functioning this way, and not other parts of the brain, then [activating mGluR5] might not be a super good treatment,” says Elizabeth Berry-Kravis, professor of pediatrics and neurological sciences at Rush University Medical Center in Chicago, Illinois, who was not involved in the study. “It might make other areas worse.”
Chattarji and his colleagues are exploring ways in which they could combine different types of drugs — some mGluR5 blockers, some activators or some that use other mechanisms — to target multiple brain areas at once.
Some researchers, including Berry-Kravis, are instead looking to target mechanisms that fall downstream of mGluR5 and may converge across brain areas.
Whether or not the new work points to new treatments, it highlights the importance of studying amygdala dysfunction in fragile X syndrome and in autism in general, says Eric Klann, director of the Center for Neural Science at New York University in New York City. Klann was not involved in the new work, but he co-authored the 2010 study with Chattarji.
“I find [the lack of research in this area] surprising and disappointing, given that the amygdala is the emotional center of the brain,” Klann says, particularly as people with fragile X syndrome and autism often display atypical emotional behaviors.
Moving forward, Chattarji and his colleagues say they plan to perform similar analyses and compare amygdala function across other animal models of autism.