This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
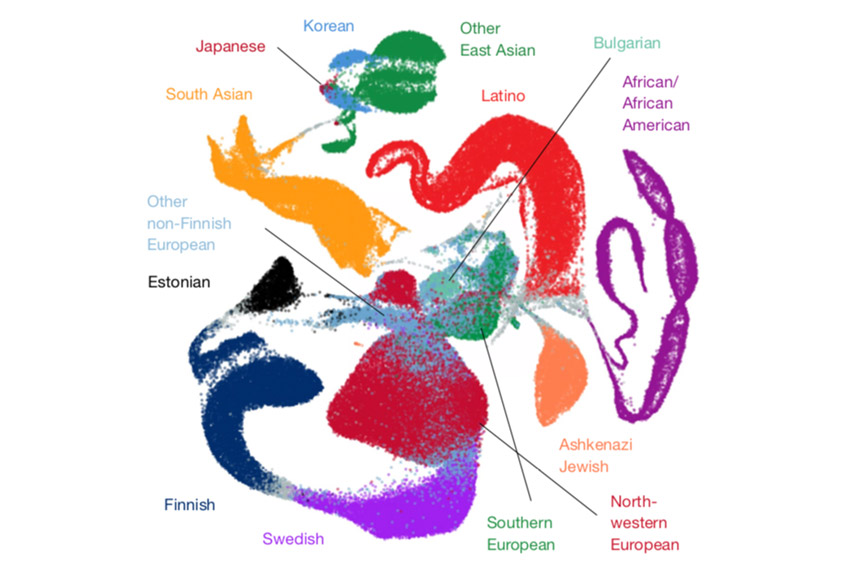
A trove of DNA sequences from 141,456 people — and counting — offers an unparalleled look at genetic variation.
A trove of DNA sequences from 141,456 people — and counting — offers researchers an unparalleled look at genetic variation across the general population1,2. The resource has been helping researchers to identify variants that contribute to autism since it was released online about four years ago3,4.
The genomes of autistic people harbor hundreds of potentially harmful mutations. But to firmly connect a specific variant to the condition, researchers need to see if it is common among typical people — a sign that that variant may actually be benign.
In 2014, researchers debuted one of the first tools to probe the prevalence of a mutation in the general population. Known as the Exome Aggregation Consortium (ExAC), it contained 60,000 sequences of exomes — the protein-coding regions of the genome5.
Two years later, the team doubled the number of exomes, incorporated whole-genome sequences and renamed the resource the Genome Aggregation Database, or gnomAD. The version the team describes in one of two new studies, published in May in Nature, catalogs 125,748 exomes and 15,708 genomes, and it continues to grow: The latest online version includes about 56,000 additional genomes, and the researchers plan to release another version at the end of 2020.
The team created the database by compiling sequences donated by more than 100 researchers, who had collected them to study conditions such as type 2 diabetes and schizophrenia, or to document genetic variation in the general population. The gnomAD team then looked for alterations at every site in the sequences, including changes to single DNA letters and ‘indels,’ which are small insertions or deletions of DNA.
The database catalogs nearly 241 million variants. It also includes sequences from individuals of African, Latino and Asian ancestries, who are often underrepresented in genetic studies.
Using the exome data, the researchers created a new way to rank each gene’s intolerance to mutations that likely compromise the function of the resulting protein. Genes that are involved in conditions such as autism tend to be intolerant of these so-called ‘loss-of-function’ variants. In more tolerant genes, by contrast, loss-of-function variants can accumulate with little effect.
The team first identified 443,769 likely loss-of-function variants in the database. For each affected gene, they calculated a score based on the number of its observed variants divided by the number that would be expected to occur by chance. They then split the genes into 10 equal groups based on this score, from lowest to highest. A low score suggests that mutations within the gene are probably harmful and less likely to propagate through the population.
The ranking is consistent with findings from other studies: Mice with a mutation in a gene that has a low score are more likely to die before birth than those with a mutation in a high-scoring gene, the researchers report.
People with intellectual disability or developmental conditions are 15 times as likely as typical individuals to have mutations in genes that are the most intolerant to variation. Similarly, autistic people are about four times as likely to have mutations in this group of genes. These findings suggest that genes’ intolerance ranking could help prioritize genes that likely play an important role in these conditions, the researchers say.
In the other study, published in the same issue of Nature, another team of researchers used the gnomAD database to catalog structural variants — DNA rearrangements that involve at least 50 DNA ‘letters.’
Structural variants account for a large part of the genetic variation among people, but scientists have mapped and sequenced relatively few of them. Sequencing often consists of piecing together short fragments of DNA, or ‘short reads,’ which miss these large rearrangements. Researchers can detect structural variants using longer reads, but it is expensive.
The researchers used a variety of algorithms to pick up on anomalies in short-read sequencing data that might flag structural variants in 14,891 genomes. They cataloged 433,371 structural variants in total — seven times more than previous studies that took a similar tack, they say.
About half of the variants are unique, meaning each appears in only one person, the researchers report. The researchers also identified 7,439 structural variants in each genome, on average, and structural variants account for about one-quarter of all rare mutations that lead to a loss of function in a gene.
The second team also measured genes’ intolerance to structural variants, similar to the measure the other team used to assess smaller mutations. A gene’s intolerance to smaller mutations correlates strongly with its intolerance to structural variants involving deletions or duplications of one of its two copies, the researchers report. This suggests that, for genes that are particularly sensitive to loss-of-function mutations, duplication can also have damaging effects.
The gnomAD database and catalog of structural variants may help researchers identify which candidate genes are most likely to be implicated in autism, the researchers say. Both resources can be accessed online in the gnomAD browser.