This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Mutations in an autism gene called ANK2 may alter brain wiring by causing the growth of excess neuronal connections.
Mutations in ANK2, a top autism gene, may alter brain wiring by causing the growth of excess neuronal connections, according to a new study in mice1.
ANK2 encodes a protein called ankyrin B. The protein comes in two main forms: a giant version found only in neurons and a smaller one found throughout the body. Mice missing both forms typically die immediately after birth, and their neuronal extensions, or axons, are unusually short.
The mice in the new study are the first to carry a mutation that impairs only the giant form of ankyrin B. They all survive and have a typical lifespan, but their axons sport a plethora of branches — an unusual development.
“Axons don’t normally form branches, which makes sense because otherwise your axon would never get to where it needs to go,” says lead investigator Vann Bennett, George B. Geller professor of biochemistry at Duke University in Durham, North Carolina.
The findings suggest that giant ankyrin B typically represses branch formation, enabling axons to reach their correct targets during brain development. And they point to a function for giant ankyrin B in the brain, previously a mystery.
“This is remarkable,” says Matthew Rasband, professor of neuroscience at Baylor College of Medicine in Houston, Texas, who was not involved in the study. “It’s the first animal model that begins to investigate how mutations in ANK2 contribute to autism.”
Bennett and his colleagues made mice with a mutation in ANK2 that resembles one identified in an autistic person. As pups, mice with this mutation make fewer squeaks than usual when separated from their mothers. As adults, they groom themselves more than controls do after being misted with water, a behavior reminiscent of the repetitive behaviors seen in autistic people.
Like the person with the mutation, the mice show no cognitive difficulties. They also have no motor problems, and the males behave like those of low social rank.
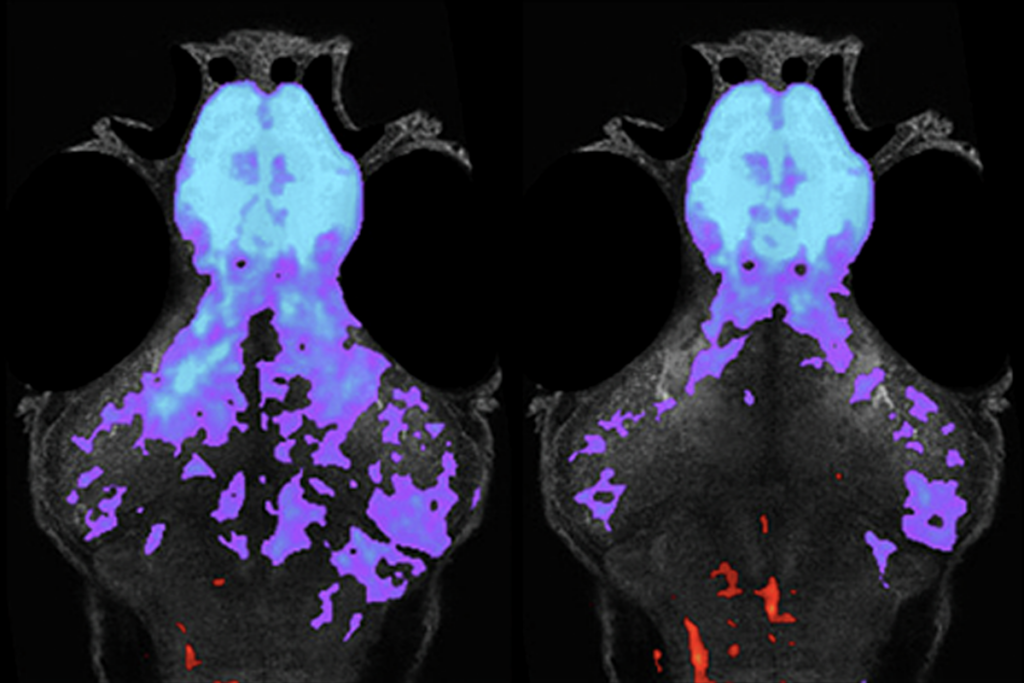
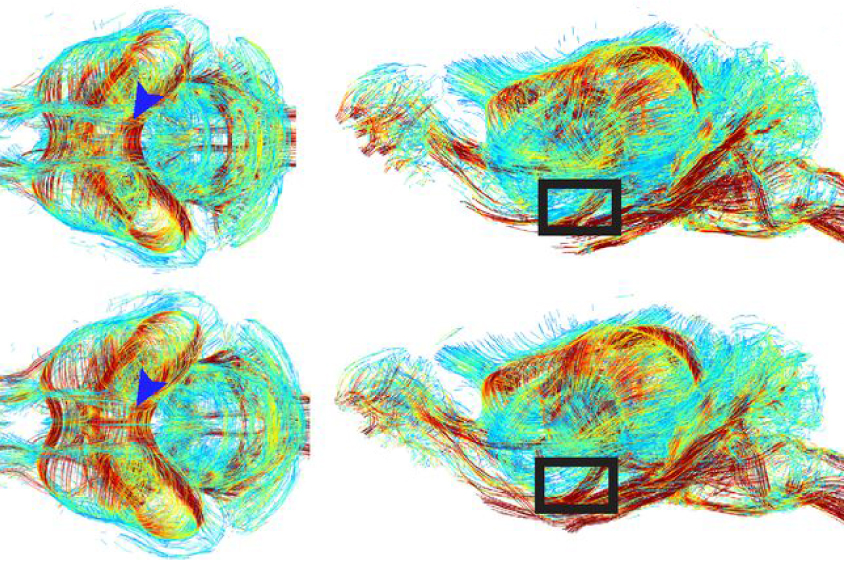
Brain slices from 28-day-old adult mutant mice show an excess of neuronal connections, or synapses, that boost neuronal activity, and — perhaps as a way to compensate — their neurons are less excitable than usual. Brain scans also show slightly more connections between pairs of brain regions than in controls. These connections are less symmetrical across the left and right halves of the brain than in controls, meaning their axons may be more randomly distributed.
Differences in synapse number and in neuron excitability are not evident in brain slices from 60-day-old mice, however, suggesting the changes are temporary. The findings appeared in July in the Proceedings of the National Academy of Sciences.
Cultured mutant neurons also form about twice as many axonal branches as control neurons do.
“It’s possible that the connectivity problems could be caused by excess axon branching,” says Noelle Dwyer, associate professor of cell biology at the University of Virginia in Charlottesville, who was not involved in the study.
Axons begin to branch by forming nubs called filopodia. Most nubs in control neurons disappear soon after they form. In the mutants, by contrast, a cell structure called a microtubule stabilizes the nub, enabling a branch to form.
The results suggest that ANK2 normally acts to block microtubules from stabilizing the nubs.
Some other autism genes, including PTEN and DYRK1A, are also known to affect neuronal branching2. And DYRK1A interacts with microtubules.
“I don’t think people have clearly investigated the idea that axon branching could be an underlying cause of autism,” Rasband says. “This is a new avenue.”
Bennett’s team is exploring how the giant ankyrin B protein interacts with microtubules.