This article is more than five years old.
Neuroscience—and science in general—is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Mice with mutations in the autism-linked gene WDFY3 have enlarged brains reminiscent of those seen in some children with autism, according to a study published 8 September in Nature Communications.
Mice with mutations in the autism-linked gene WDFY3 have enlarged brains reminiscent of those seen in some children with autism, finds a study published 8 September in Nature Communications1. The brain overgrowth begins in the womb, the study found.
WDFY3 plays a role in autophagy, a process that rids cells of damaged or unneeded parts. A 2012 study first identified a WDFY3 mutation in a child with autism2. The gene has since turned up in several lists of autism risk genes.
“We wanted to see if there are similar developmental anomalies in these mice that may have also been reported within the autism population,” says lead researcher Konstantinos Zarbalis, assistant professor of pathology and laboratory medicine at the University of California, Davis.
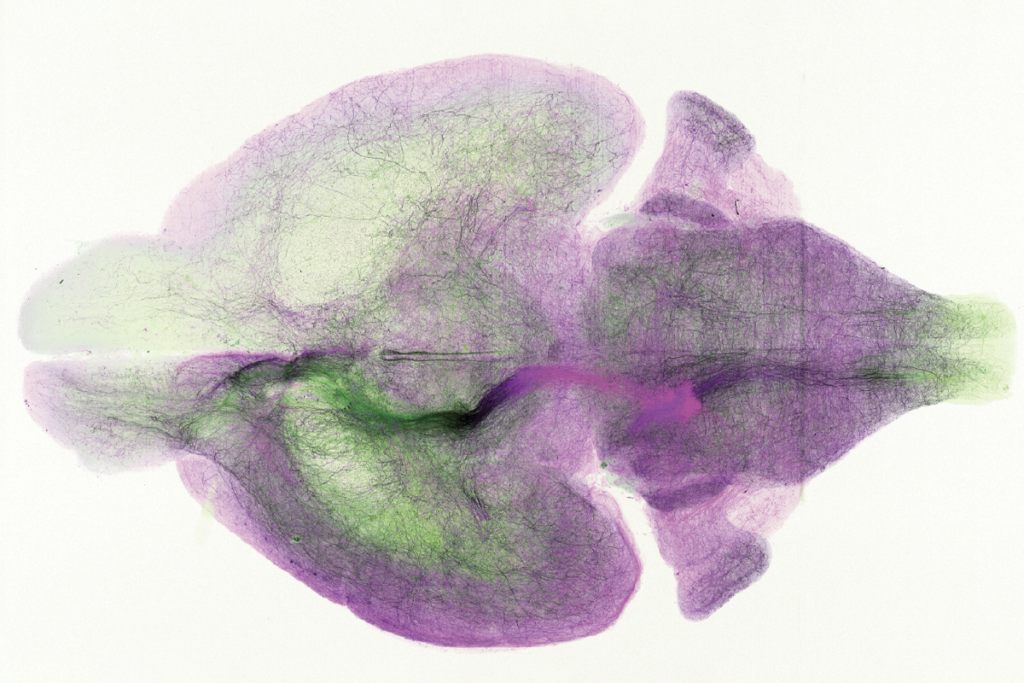
Mouse embryos with two copies of the mutant gene have enlarged brains and an excess of immature neurons that divide faster than usual. They also have misdirected patches of neurons.
Similar changes have been observed in people with autism. Some children with the disorder exhibit accelerated brain growth during the first year of life3. The resulting enlargement appears to stem from an excess of neurons, and is particularly pronounced in the frontal and temporal regions of the cerebral cortex, which are involved in social behavior and communication. The rapid growth begins to slow around age 2, and adults with autism have brains of normal size4.
“It’s really pretty fascinating,” says Eric Courchesne, professor of neuroscience at the University of California, San Diego, who was not involved in the study. “A single gene defect is producing multiple well-characterized autism attributes, all prenatally.” He adds that it is rare to see a genetic animal model of autism that captures so many autism features.
The new study supports the notion that brain overgrowth in autism may originate in the womb. “It’s really additional evidence telling us that many cases of autism, especially those that involve mutations, come back to fetal and embryonic stages of development,” says Robert Hevner, professor of neurological surgery and pathology at Seattle Children’s Research Institute in Washington.
Zarbalis says his team “stumbled” on the finding during experiments to discover genes involved in brain development. The researchers were searching for brain abnormalities in mice treated with a mutagen to generate random mutations and found a strain that shows a lack of connectivity between the cortex and other areas of the brain5. DNA sequencing of these mice revealed a single base-pair change in one copy of WDFY3.
“A single gene defect is producing multiple well-characterized autism attributes, all prenatally.”
The researchers found the mice have no detectable brain abnormalities. They then turned their attention to mice with two copies of the mutant gene, but those pups died at birth. Because of this, the researchers examined the brains of embryos and newborn pups.
“It’s important to keep in mind that they were not able to study the final mature state of the cortex,” Hevner notes. “I would say this would be comparable to looking at a human fetus rather than a 2-year-old child.”
The researchers found that mice with mutant WDFY3 have enlarged brains overall. Interestingly, the frontal region of the cortex in these animals is longer but thinner than the cortex in normal mice. This stems from an increased number of radial glial cells, which are progenitor cells that give rise to neurons.
The radial glial cells divide faster in the mutant mice than in controls. They are also more likely than those of controls to divide symmetrically, meaning that they produce two radial glia instead of one radial glial cell and one cell that is closer to becoming a neuron. This shift toward symmetrical divisions causes the cortex to expand in length rather than in thickness6.
The findings are in line with those from a 2011 study that found children with autism have too many neurons in the frontal region of the cortex7. New neurons in the human brain are made primarily during embryonic and fetal development.
The mutant mouse brains also have abnormal patches containing cells that did not migrate to their proper destination. Researchers have observed similar patches in the postmortem brains of children with autism8.
“It’s quite striking,” Courchesne says. “It suggests that a lot of these anatomic findings in [children with autism] could be generated by genetic factors that are playing out early in development.”
To confirm that these characteristics are the result of loss of WDFY3 function, Zarbalis and his team engineered mice with a large deletion in both copies of WDFY3. These animals have even bigger brains containing more abnormal patches than do the original mutants, but they also die before birth.
It’s still unclear how the loss of WDFY3 function leads to these brain abnormalities. The researchers found no evidence for disruptions in autophagy, despite the gene’s known role in the process. “It suggests that WDFY3 may have some other important biological function,” Hevner says.
Zarbalis plans to examine the effects of deleting WDFY3 from only certain brain regions in the hopes that this will help the animals survive past birth. “We hope to see in these mice behavioral abnormalities that reflect aspects of the autism behavioral core symptoms,” Zarbalis says.
1. Orosco L.A. et al. Nat. Commun. 5, 4692 (2014) PubMed
2. Iossifov I. et al. Neuron 74, 285-299 (2012) PubMed
3. Courchesne E. et al. Neurology 57, 245-254 (2001) PubMed
4. Hazlett H.C. et al. Arch. Gen. Psychiatry 68, 467-476 (2011) PubMed
5. Zarbalis K. et al. PLoS Biol. 2, E219 (2004) PubMed
6. Fish J.L. et al. J. Cell Sci. 121, 2783-2793 (2008) PubMed
7. Courchesne E. et al. JAMA 306, 2001-2010 (2011) PubMed
8. Stoner R. et al. N. Engl. J. Med. 370, 1209-1219 (2014) PubMed